综述|全面了解比线粒体更热的内质网自噬!
ER-phagy: mechanisms, regulation, and diseases connected to the lysosomal clearance of the endoplasmic reticulum
Fulvio Reggiori and Maurizio Molinari
17 MAY 2022https://doi.org/10.1152/physrev.00038.2021
我们都知道自噬是细胞通过溶酶体或液泡(酵母),将受损或多余的细胞组分降解产生能量或生物小分子的代谢过程,在真核生物中具有高度保守性,与人类健康密切相关。在过去的25年里,大量开创性的发现使细胞自噬成为生命科学和医学领域的一个重要研究领域,但是关于内质网(ER)自噬的机制探讨并不深入,相关高分研究类文章更是屈指可数。到底是什么阻碍了对ER自噬的研究步伐?让我们先通过一篇发表于Physiological reviews(IF:46.500)的综述文章,大概了解一下ER自噬及其生理病理的分子机制。
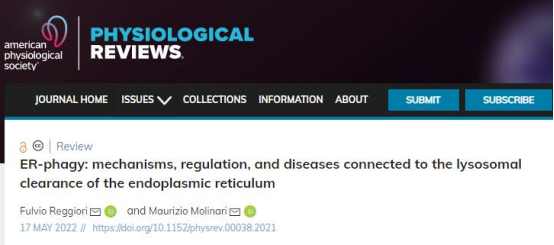
ER自噬是如何发生的?ER自噬反应是由大量嵌入ER膜或被募集到ER膜上的ER自噬受体协同或个体激活所驱动。当蛋白质或蛋白质复合物、入侵的病原体或单个细胞器(如线粒体或ER)等“吃我信号”受损时,大量自噬和自噬受体介导的程序被激活,ER部分片段化后递送至溶酶体/液泡区室进行清除,用以消化细胞质内容物。这一过程主要通过ER巨自噬、ER微自噬及LC3依赖性囊泡三种途径驱动蛋白质、膜和糖的快速动员以生成构件,调节细胞主要生物合成细胞器的大小、形状和活性(Fig. 1)。
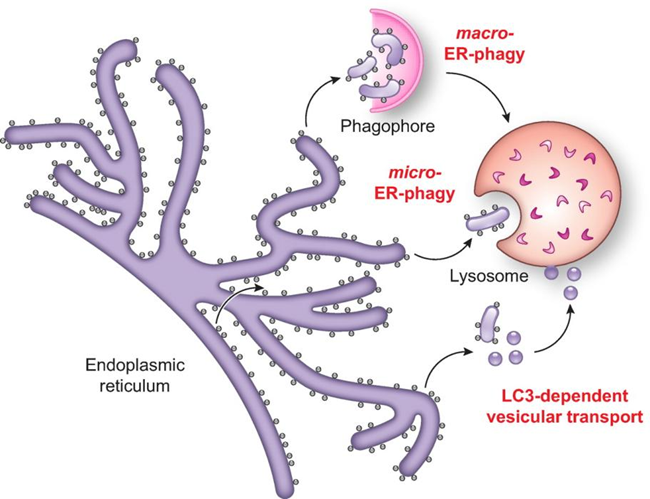
Fig. 1 ER自噬的三个主要机制哪些ER自噬受体参与其中?文章中作者描述了哺乳动物、酵母以及植物ER自噬受体,这里选取与人类更接近的哺乳动物部分进行介绍(Fig. 2)。细胞溶质Atg8/LC3结合蛋白最近已被证明仅在ER自噬激活时被募集到ER膜上,并且可以作为ER自噬受体发挥作用。激活后,膜包埋和可溶性ER自噬受体与Atg8/LC3蛋白家族成员以及调节自消化程序的管腔、胞质蛋白和功能复合物结合。膜结合的ER自噬受体FAM134B最初被定义为肿瘤抑制基因并命名为JK1,最近研究通过酵母双杂交筛选将其鉴定为Atg8/LC3蛋白的互作蛋白,神经元细胞在FAM134B缺失后出现ER肿胀。SEC62控制新合成蛋白到ER的转运,在营养剥夺被激活时参与ER巨自噬,在ER应激恢复期间被激活时参与ER微自噬。网状细胞(RTN)蛋白富含管状ER,通过在营养剥夺细胞中的过表达分析,发现只有RTN3L(RTN3的大同种型)诱导ER片段化以及ER和Atg8/LC3蛋白之间的共定位,促进饥饿诱导的内质网小管溶酶体清除。单一II型跨膜内质网蛋白—细胞周期进程基因1(CCPG1)作为ER应激诱导基因,通过Atg8/LC3蛋白和FIP200的相互作用,参与饥饿诱导的ER自噬。
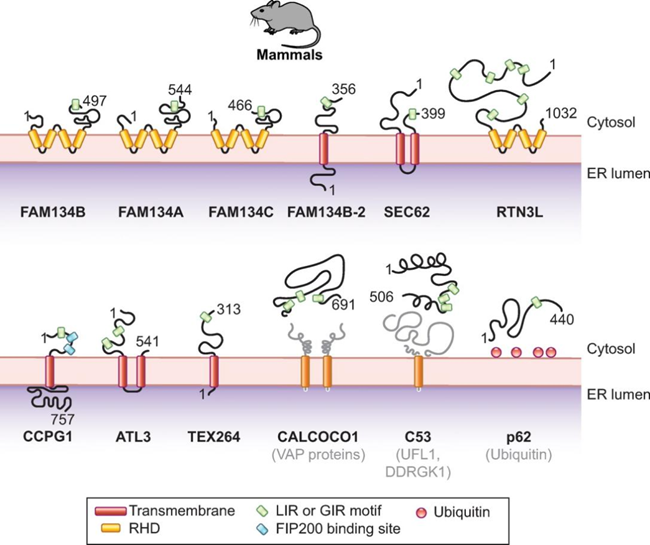
Fig.2 哺乳动物ER自噬受体胞质ER自噬受体CALCOCO1与TAXBP1参与高尔基体亚域的溶酶体转换;当ER自噬是由在错误mRNA上停滞的核糖体产生的新生蛋白堵塞而诱导时,C53开始发挥作用;p62通过识别ER膜蛋白促进泛素化底物的自噬清除。ER部分的划分和片段化ER部分的分离和碎片化是ER自噬途径中必不可少的过程,但具体定义尚不明确。大多数ER自噬受体具有非常短的氨基酸延伸,突出到ER腔中(可溶性受体除外),依赖于与其他内质网蛋白的结合来感知内质网腔中的扰动和/或标记编码“吃我”信号的膜部分。如FAM134蛋白家族成员与凝集素伴侣CNX的关联,其与错误折叠蛋白质的持续结合可以将它们分离在ER子域中,这些子域最终形成小泡并通过ER到溶酶体相关的降解途径与其内容物一起降解。ATL因具有细胞溶质动力蛋白样GTP酶结构域,成为主动操控内质网膜断裂的最佳候选蛋白质,ATL2的缺失会抑制FAM134B调节的营养缺乏细胞中ER层在溶酶体的降解。触发ER自噬的因素触发ER自噬反应的信号源自ER内部,包括从ER应激中恢复、错误折叠的多肽积累、核糖体停滞和病原体入侵。它们引发选定ER部分的溶酶体降解,留下不受影响的细胞质大分子和其他细胞器。营养缺乏在细胞外营养物质短缺的情况下,细胞存活依赖于自噬程序的激活,其中部分细胞质内容物在溶酶体/液泡内降解为基本代谢物,从而产生内部营养物质库。需要注意的是,大量的代谢物被认为是营养物质,这里描述的“营养缺乏”并未具体说明细胞或生物体到底缺少什么。对于哺乳动物来说,研究人员主要通过剥夺细胞的葡萄糖、氨基酸或血清(或联合剥夺)来研究自噬,这或许也是导致自噬调节和/或响应营养限制的进展有差异的原因(Fig. 3)。
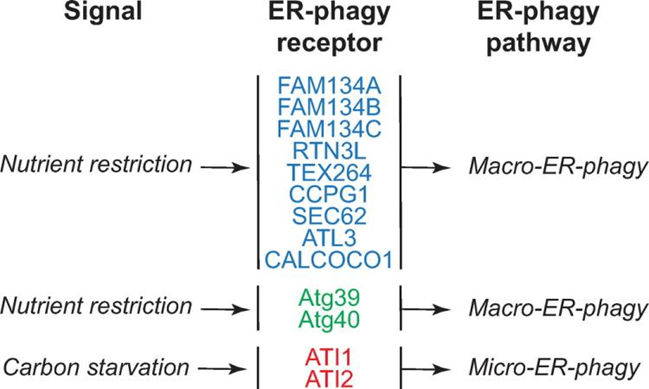
Fig.3 激活ER自噬反应的信号病理性细胞应激细胞稳态的扰动会激活一组称为ER应激传感器的ER膜蛋白的翻译后修饰,这里的翻译后修饰主要指IRE1蛋白的寡聚化和磷酸化以及ATF6蛋白和PERK的蛋白水解加工,可由多效生理信号触发,如细胞分化,也可由病理信号触发,如病原体感染、干扰蛋白质N-糖基化、钙或氧化还原稳态的化合物,以及错误折叠蛋白质的积累(Fig. 4)。
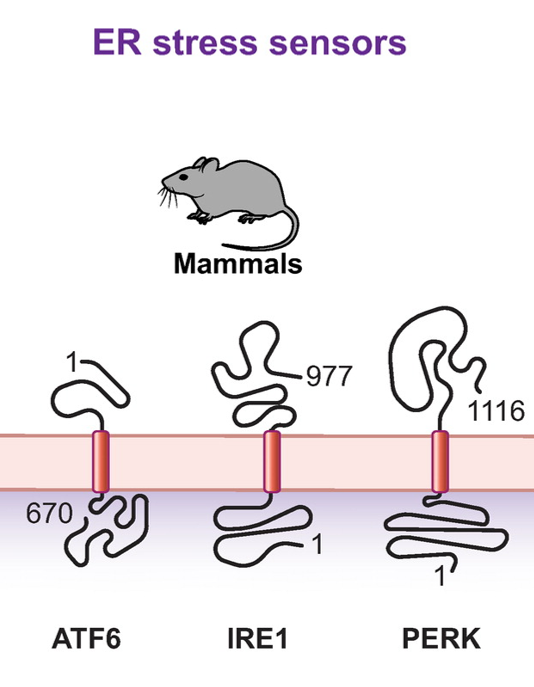
Fig.4 ER应激传感器ER应激传感器的激活会触发下游信号特异性转录和翻译程序,这些未折叠的蛋白质反应最终会增加ER大小、ER驻留蛋白质水平及整体ER活性。分解代谢ER自噬反应和合成代谢未折叠蛋白反应的协同活动确保了ER的可塑性,以快速适应不断变化的稳态需求。在一项实验中,中断对白化大鼠的抗癫痫药物苯巴比妥药物治疗后,钙或氧化还原稳态的扰动会引起称为ER应激恢复的ER自噬,以去除多余的ER,这种恢复性ER自噬也受SEC62调节,但与多效信号引起的ER自噬不同(Fig. 5)。
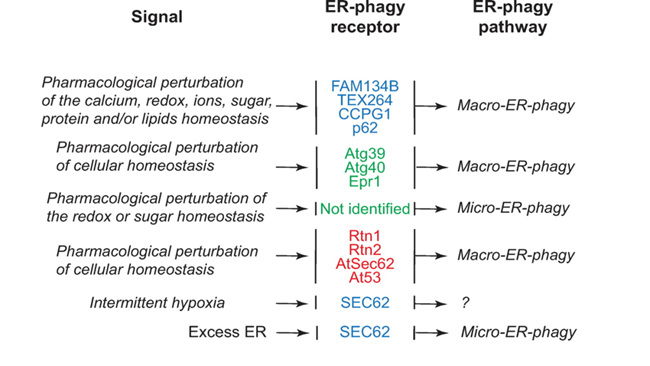
Fig.5 激活ER自噬反应的信号错误折叠蛋白核糖体停滞、多肽形成受阻、病原体入侵都会触发ER应激的ER自噬。错误折叠的蛋白质须迅速从ER中被去除以避免细胞毒性,大多数错误折叠蛋白或穿过ER膜,或从ER膜中被提取进行多泛素化,最后被细胞溶质蛋白酶体降解。另外少数错误折叠多肽隔离在特定ER子域中,与ER自噬受体结合激活ER自噬,促进这些ER部分的囊泡形成及溶酶体降解(Fig. 6)。另一种模型认为少数错误折叠的多肽被转运穿过内质网膜到达细胞质形成自噬体从而被隔离,通过巨自噬被消除。有趣的是,最近有报道称在神经突生长期间,ER自噬机制的特定成分参与了一种途径,该途径不会导致ER溶酶体清除,但会导致其从细胞中被排出。
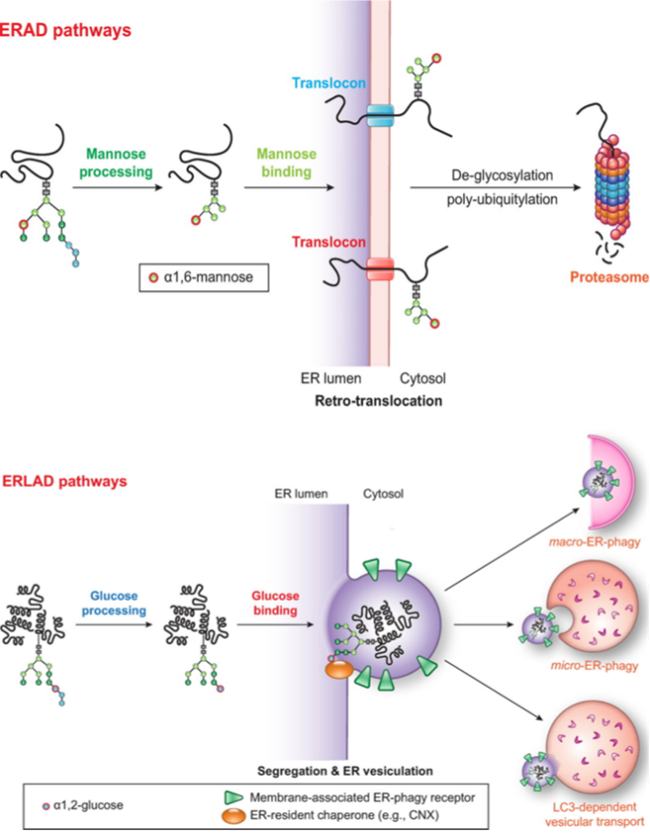
Fig.6 ER降解ER自噬与人类相关疾病ER自噬受体的基因突变与许多疾病有关,最新数据表明,其中一些可能是病毒和细菌病原体的目标(Table. 1)。神经系统疾病如FAM134B的突变导致遗传性感觉和自主神经病2B(HSAN2B),这是一种常染色体隐性遗传病,其特征是有髓和无髓纤维的缺失。尽管病理生理学上HSAN2B与ER自噬缺陷没有直接关系,但在部分患者中发现Glu145截断移除了部分网状同源结构域和LIR基序,从而导致FAM134B在ER的碎裂,随后被递送至溶酶体。RTN3L在神经元中含量丰富,与阿尔茨海默病有关,在大脑中,β淀粉样蛋白转化酶1(BACE1)主要与神经元中的RTN3共定位,任何RTN蛋白表达的增加都会大大减少Abeta的产生。拓扑异构酶1(TOP1)与DNA之间催化中间体的稳定化形成的高度细胞毒性复合物(TOP1裂解复合物)会引发基因组不稳定,是神经系统疾病的诱因之一。研究发现TEX264是修复TOP1裂解复合物的核心参与者,在面向胞质溶胶(用于ER自噬)的ER和核膜或面向细胞核(用于DNA修复)的内核膜上充当ER自噬受体,可能有助于协调核噬过程中核内成分的选择性降解从而维持基因组稳定性。肿瘤在结肠癌(CRC)细胞中,已发现FAM134B与EB1/MAPRE2互作,EB1/MAPRE2是微管细胞骨架组织中的关键,通过与微管的正端结合并作为多种相互作用蛋白的平台,控制微管动力学。TEX264也是CRC的标记蛋白,并且与FAM134B一样,在暴露于药物治疗诱导的内质网应激和自噬诱导的细胞死亡的胶质母细胞瘤细胞中,TEX264被强烈诱导。CALCOCO1的表达也被发现在CRC和乳腺癌中发生了改变。病原体感染感染登革热病毒、寨卡病毒和西尼罗河病毒的细胞会导致FAM134B的裂解,但不会导致其它ER驻留蛋白(包括ATL2和ATL3)裂解,这些病毒表达的NS3蛋白酶与FAM134B共定位,且其NS2B辅助因子是FAM134B特异性加工所必需的,产生的片段无法维持ER自噬。FAM134B的减少会促进登革热和寨卡病毒的感染。过敏性鼻炎该疾病是由各种白细胞的复杂相互作用驱动的,疾病风险已关联到上位相互作用蛋白FAM134B。
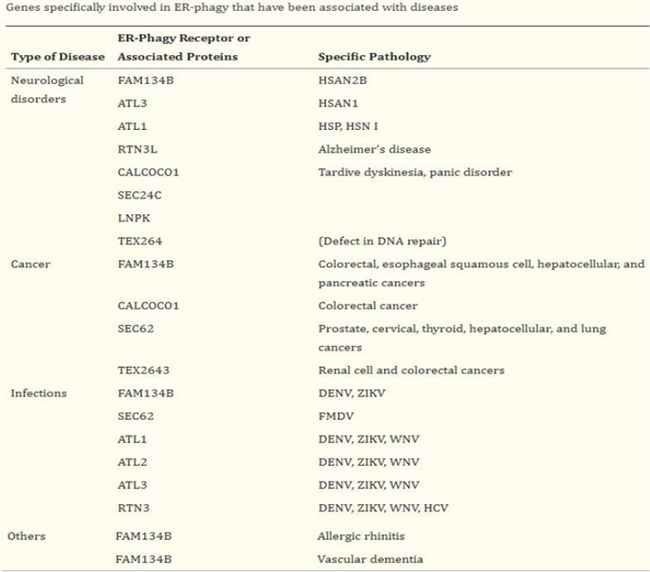
Table.1 与疾病相关的ER自噬基因
小编总结虽然参与ER自噬的分子和诱导ER自噬发生的机制研究已取得巨大进展,但是不同的生理和病理信号是如何参与特定的ER自噬机制以及ER片段化和随后隔离到自噬体、溶酶体和/或核内体中的精确机制仍是亟待回答的核心问题。ER自噬为饥饿细胞提供营养、选择性诱导以消除功能失调和多余的ER、参与细胞发育和分化,依赖于ER自噬反应及相关基因突变引起相关疾病的生理病理学过程有待被发现,揭示和阐明更多的ER自噬机制才能提高治疗这些疾病的可能性。

