Endoplasmic reticulum stress signals in the tumour and its microenvironment

内质网(ER)中蛋白质的处理、修饰和折叠是一个受到严格调控的过程,决定着细胞的功能、命运和生存。在一些肿瘤类型中,不同的致癌、转录和代谢异常共同产生不利的微环境,破坏恶性细胞和基质细胞以及浸润性白细胞的内质网稳态。这些变化引发了持续的内质网应激状态,已被证明在对先天和适应性免疫细胞的功能进行动态重编程的同时,可以控制癌细胞中的多种促肿瘤属性。因此,内质网应激传感器及其下游信号通路的异常激活已成为肿瘤生长和转移以及对化疗、靶向治疗和免疫治疗反应的关键调节因子。
内质网 (ER) 是一个中央细胞器,分泌蛋白和跨膜蛋白在这里合成、折叠和修饰。尽管这个过程受到精确调控,但多种外部因素和细胞内在事件会破坏该细胞器的蛋白质折叠能力,并引发内质网应激状态,其特征是错误折叠或未折叠蛋白质的积累。肿瘤中富集的多种遗传、转录和代谢异常产生不利的微环境,导致肿瘤细胞持续存在 ER 应激,最终影响它们的功能、命运和存活(图 1)。
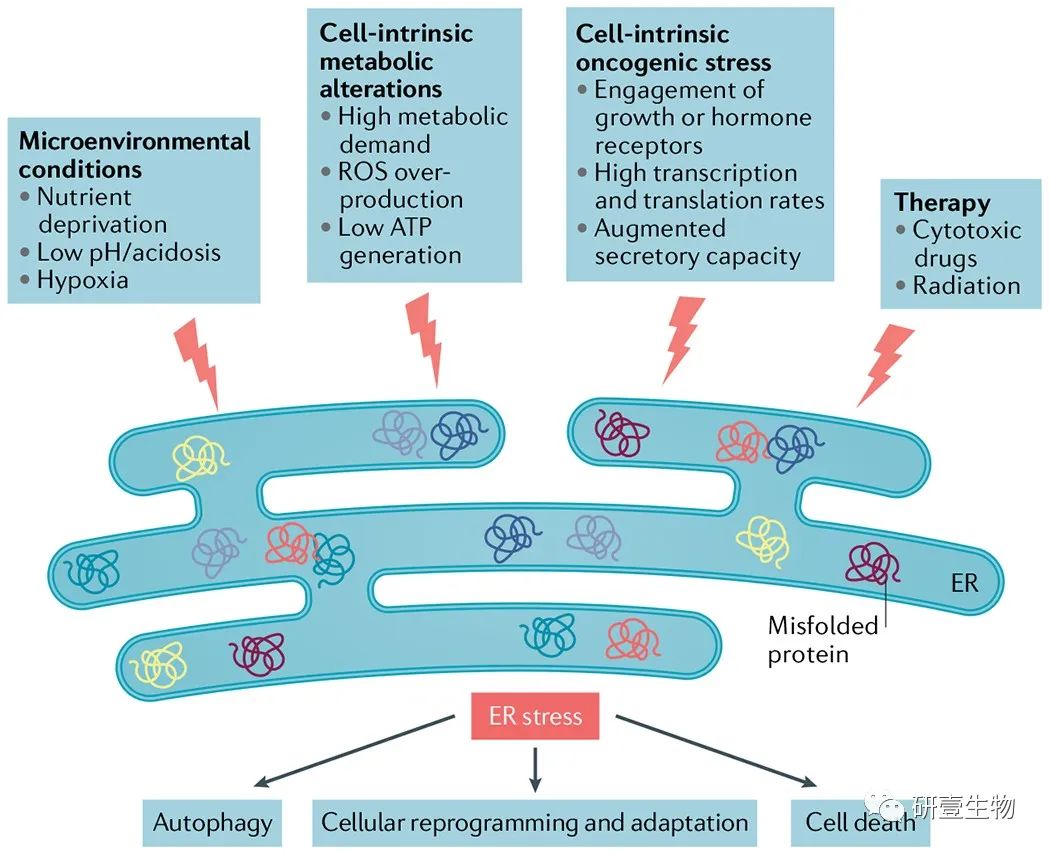
图 1
肿瘤微环境中内质网应激的诱导剂。
肿瘤生长中恶性细胞不受控制的增殖能力会产生不利的微环境,其特征是高代谢需求、缺氧、营养限制和酸中毒,这反过来又会破坏居住在这种环境中的多种细胞类型的钙和脂质稳态。总的来说,这些严酷的条件改变了癌细胞和浸润性免疫细胞中内质网 (ER) 的蛋白质折叠能力,从而促进了错误折叠或未折叠蛋白质在该细胞器内的积累,从而导致 ER 应激。癌细胞中的致癌事件通过提高它们的全局转录和翻译率进一步促进了这种状态。未折叠蛋白反应 (UPR) 随后被激活,试图恢复 ER 稳态并促进适应肿瘤中的各种损伤。某些治疗方式还可以触发癌细胞中的内质网应激,从而改变它们在肿瘤微环境 (TME) 中的正常行为。根据内质网应激的程度、细胞类型和特定的病理背景,内质网应激反应可以产生多种影响,从细胞重编程和适应到自噬和细胞凋亡。由于在癌症起始、进展和治疗过程中同时富集在 TME 中的各种 ER 应激源的累加效应,强烈和持续的 UPR 激活主要在体内的癌细胞和肿瘤浸润免疫细胞中得到证实,这在体外条件下一直具有挑战性。
协调 ER 应激反应是一个高度动态的过程,可能导致促生存和促凋亡产出。事实上,细胞命运的决定似乎取决于 UPR 的强度和持续时间(图 2)。在过去的 15 年中,多项研究揭示了 ER 应激反应通路在癌症发生和发展中的相关作用。
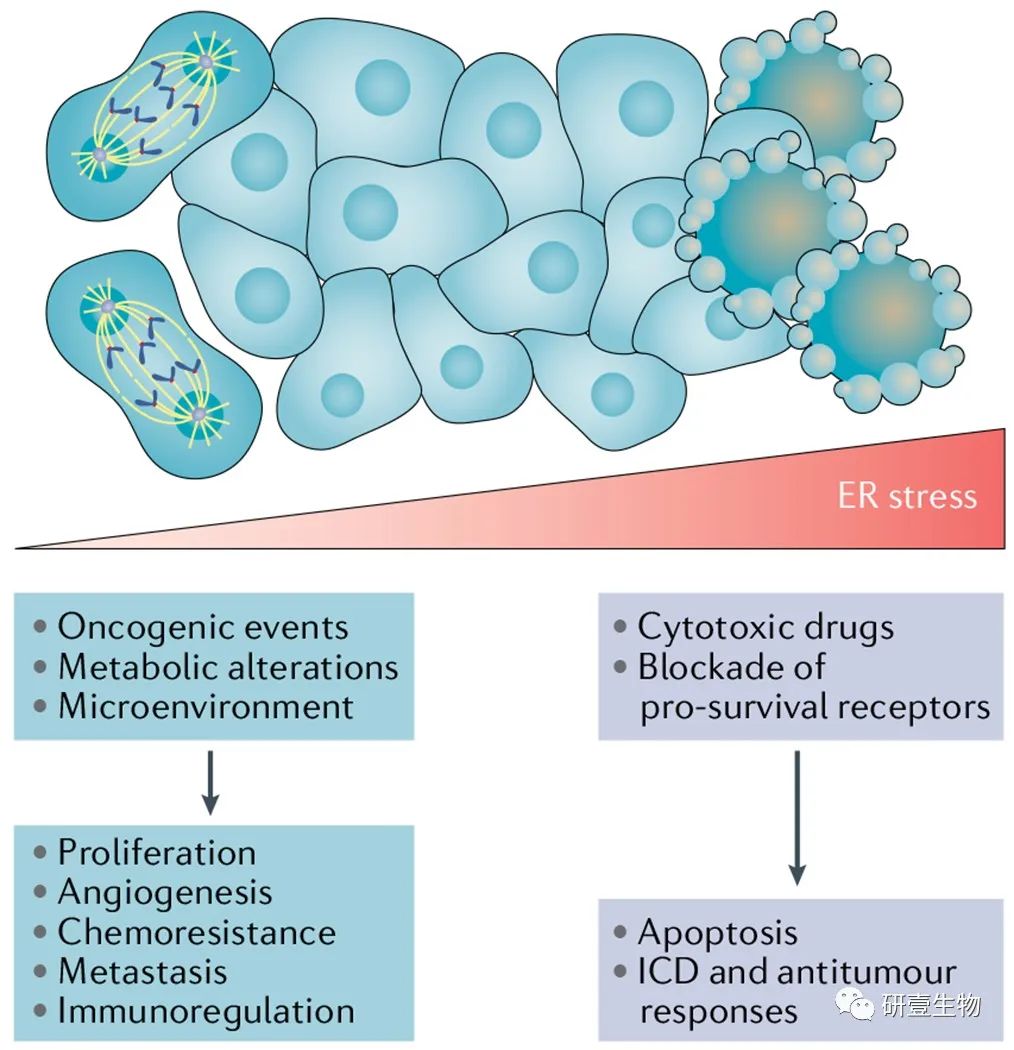
图 2
内质网应激的大小及其在恶性细胞中的不同结果。
由致癌途径、代谢变化和肿瘤微环境条件引发的持续但适度的内质网 (ER) 应激反应刺激了几种促进癌细胞增殖、转移、化疗耐药、血管生成和免疫逃避的机制。相比之下,由错误折叠蛋白在该细胞器中不受控制的积累引起的极端 ER 应激可导致终末未折叠蛋白反应 (UPR)。从而诱导细胞死亡。例如,蛋白酶体抑制剂已被证明可通过过度激活 PRKR 样 ER 激酶 (PERK)–真核翻译起始因子 2α (eIF2α)–激活转录因子 4 (ATF4)–C/EBP 来触发多发性骨髓瘤细胞中的促凋亡 ER 应激反应UPR 的同源蛋白 (CHOP) 臂。值得注意的是,暴露在一些细胞毒性药物,如蒽环类药物,可触发内质网应激反应,促进能够引发抗肿瘤免疫的免疫原性细胞死亡(ICD) (box2)。因此,UPR 激活的结果,无论是促生存还是促凋亡,都取决于应激的持续时间和强度。
致癌转化是一个多步骤过程,利用普遍定期审议来克服各种障碍。因此,完整的 UPR 对于适应由 MYC 驱动的致癌转化引起的压力至关重要(图 3)。
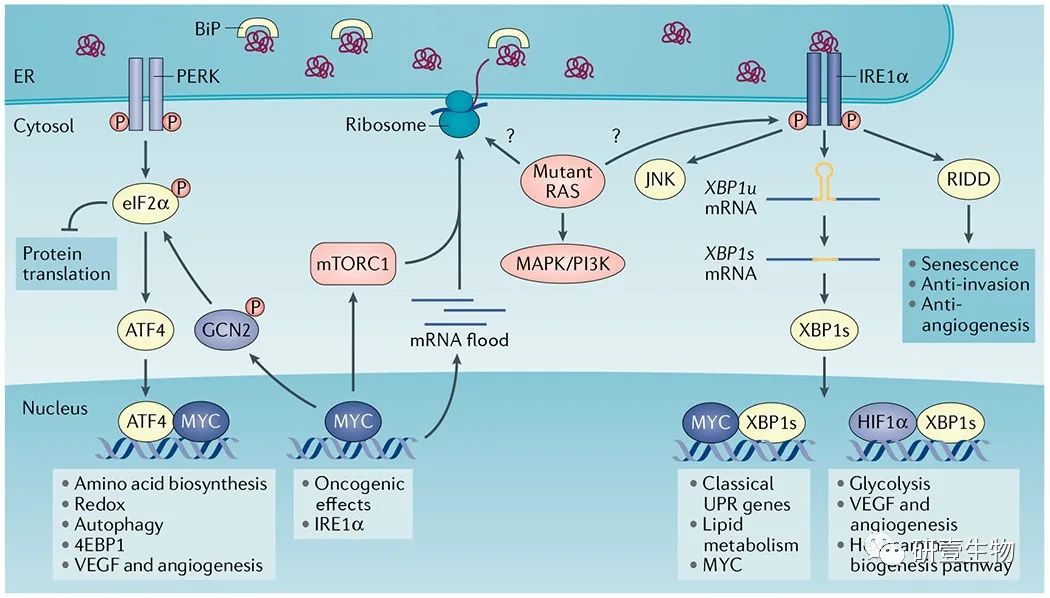
图 3
癌细胞中致癌程序和内质网应激反应的整合。
致癌性 MYC 通过多种机制激活未折叠蛋白反应 (UPR)。MYC 诱导的全局转录上调(mRNA 泛滥)和翻译增加了内质网 (ER) 中的核糖体生物发生和蛋白质负荷,从而激活了 UPR 的所有分支。MYC 进一步结合编码肌醇需要蛋白 1α (IRE1α) 的基因中的启动子和增强子区域,正向调节其转录并提高 IRE1α 蛋白水平。MYC 还可以在细胞核中与 X-box 结合蛋白 1s (XBP1s) 形成异源二聚体,以调节经典的 UPR 基因和脂质代谢基因。值得注意的是,XBP1s 已被证明可以促进前列腺癌细胞和自然杀伤细胞中的 MYC 转录。MYC 参与 PRKR 样 ER 激酶 (PERK) 和一般性调控阻遏蛋白激酶2 (GCN2),以诱导真核翻译起始因子 2α (eIF2α) 磷酸化和综合应激反应。MYC 还可以与活化转录因子4 (ATF4) 相互作用,以调节氨基酸转运体和生物合成、抗氧化途径和自噬。MYC-ATF4 复合物调节真核翻译起始因子 4E 结合蛋白 1 (4EBP1),以减少翻译和蛋白毒性应激。mTOR 复合物 1 (mTORC1) 激活诱导蛋白质合成和激活 UPR 的 ER 超载。反过来,IRE1α-肿瘤坏死因子受体相关因子 2 (TRAF2)-JUN N-末端激酶 (JNK)-胰岛素受体底物 1 (IRS1) 轴已被证明限制 mTORC1 活性。突变 RAS 以特定于上下特定的方式集成在 UPR 中。突变体 HRAS 通过未知机制优先诱导角质形成细胞中的 IRE1α 活性。在原代人黑素细胞中,HRAS-G12V–PI3K 而不是 BRAF-V600E 会增加 ER 含量并诱导所有 UPR 分支的激活。目前尚不清楚突变 RAS 是否会增强 ER 中的整体蛋白质翻译和蛋白质负荷,从而促进所有癌症类型的 ER 应激。
大量研究表明,癌细胞固有的 ER 应激反应可以通过改变 TME 中共存的免疫细胞的功能来影响恶性进展(图 4)。
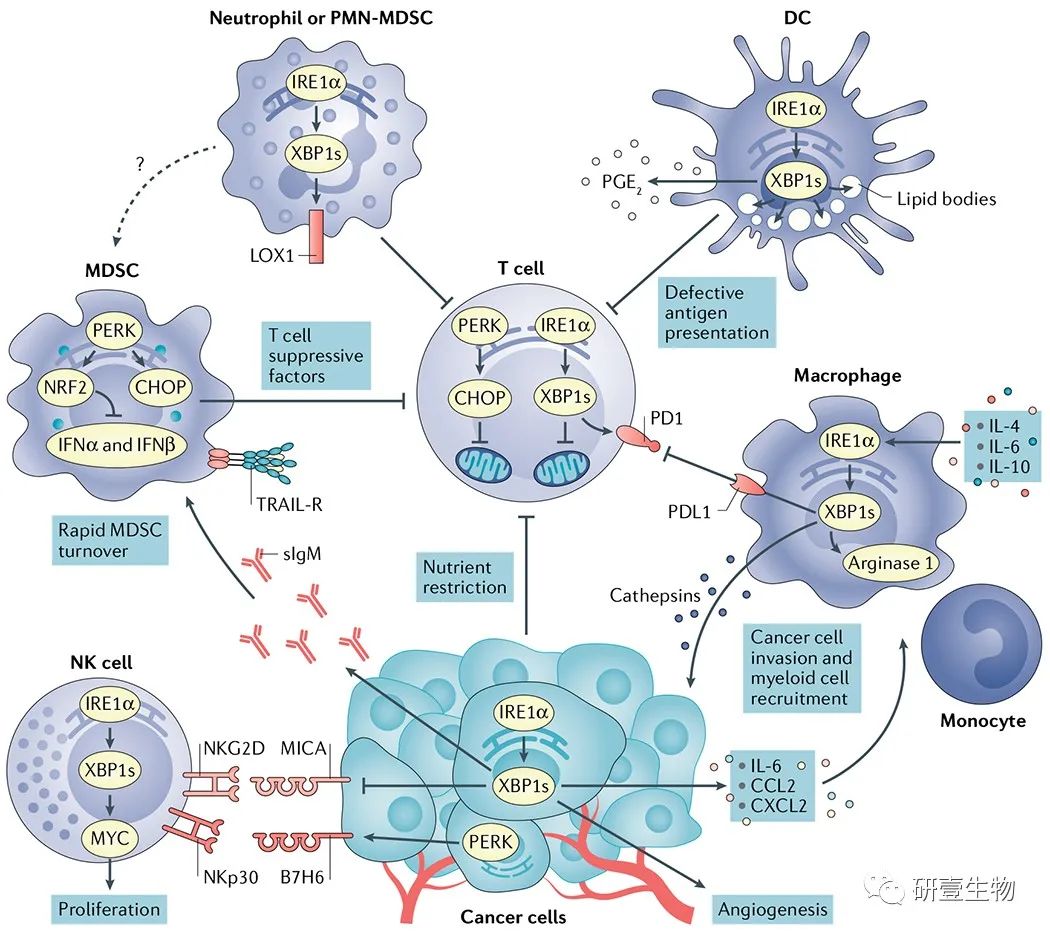
图 4
肿瘤微环境中内质网应激信号的免疫调节作用。
肌醇需求蛋白 1α (IRE1α) 或 PRKR 样 ER 激酶 (PERK) 激活的癌细胞通过自然杀伤 (NK) 细胞调节对肿瘤识别,同时分泌促进血管生成和骨髓细胞类型募集到肿瘤部位的介质。IRE1α 和 PERK 都可以很好地调节血管生成。X-box 结合蛋白 1s (XBP1s) 和激活转录因子 4 (ATF4) 直接结合血管内皮生长因子 ( VEGF)启动子来调节其表达。营养限制、活性氧(ROS)积累或抑制葡萄糖摄取的可溶性因子的存在导致肿瘤内T细胞内质网(ER)应激和未折叠蛋白反应(UPR)的IRE1α-XBP1和PERK-C /EBP同源蛋白(CHOP)臂的慢性激活,引起线粒体功能障碍并抑制其最佳抗癌效应功能。肿瘤微环境 (TME) 中高水平的胆固醇也可以激活肿瘤内 T 细胞中的 IRE1α–XBP1 信号,诱导程序性细胞死亡蛋白 1 (PD1) 表达并限制其保护活性。髓源性抑制细胞 (MDSCs) 利用 PERK 通过 CHOP 介导的 T 细胞抑制因子表达和诱导核因子红细胞 2 相关因子 2 (NRF2) 驱动的反应来控制抗肿瘤免疫,从而抑制保护性 I 型干扰素的产生。MDSCs 中的 ER 应激也与其肿瘤坏死因子相关的凋亡诱导配体受体 (TRAIL-Rs) 表达升高和 TME 中的快速更新有关。ER 应激相关基因特征和凝集素型氧化 LDL 受体 1 (LOX1) 的表达可区分癌症患者的正常中性粒细胞和多形核 (PMN)-MDSC。此外,ER 应激的中性粒细胞获得免疫抑制特性并通过 IRE1α 激活过度表达 LOX1。ROS 积累会促进肿瘤相关树突状细胞 (DC) 中的 ER 应激和持续的 IRE1α–XBP1 激活,驱动不受控制的脂滴形成,从而抑制它们将局部抗原呈递给肿瘤内 T 细胞的能力。ER 应激的 DC 也被证明会过度产生免疫抑制性脂质介质前列腺素 E2 (PGE 2 ) 通过 IRE1α–XBP1 激活,可能有助于癌症的免疫逃逸。在巨噬细胞中,IRE1α–XBP1 分支已被证明可促进组织蛋白酶、PD1 配体 1 (PDL1) 和精氨酸酶 1 的表达,进一步促进 TME 中的癌细胞侵袭和免疫抑制。
持续的 ER 应激是癌症的一个新兴标志,它是由 TME 中的多种代谢和致癌异常引起的,这些异常扰乱了恶性细胞和浸润性免疫细胞中的蛋白质折叠稳态。组成活性 ER 应激反应使恶性细胞能够适应致癌和环境挑战,同时协调促进恶性进展的多种免疫调节机制。未来应该对 UPR 对 TME 中单个细胞类型的精确影响进行系统研究,尤其是对体内癌细胞代谢重编程的影响。尽管多项研究阐明了UPR在肿瘤发生的每个步骤中的功能和机制,还需要更多的研究来了解内质网应激在癌症转移和治疗抵抗中的作用。特别是,在转移级联的限速步骤中对 UPR 的更深入的机制理解,以及在转移期间 TME 重编程的背景下,对于合理设计有效的治疗干预措施以解决当前的临床挑战并改善患者预后。

