
线粒体几乎存在于人体的每个细胞中。它负责产生维持生命和支持器官功能所需的 90% 能量。
当线粒体无法将食物和氧气转化为维持生命的能量时,细胞损伤甚至细胞死亡就会随之而来。当这个过程在全身重复时,器官系统开始衰竭,甚至停止运作。还有很多东西有待发现,但这就是我们所知道的。线粒体疾病是一种遗传性疾病。线粒体也可能受到其他遗传疾病和环境因素的影响。生物学和医学的传统教学认为线粒体仅充当细胞的“能量工厂”。这种过度简化是一个错误,它减缓了我们理解线粒体疾病生物学的进展。
制造一个线粒体需要大约 3,000 个基因。线粒体 DNA 只编码其中 37 个基因。其余的基因在细胞核中编码,所得蛋白质被转运到线粒体。这意味着制造线粒体所需的基因中只有约 3%(3000 个中的 100 个)被分配用于制造 ATP,而超过 95%(3000 个中的 2900 个)涉及与分化细胞的专门职责相关的其他功能它所在的位置。
随着我们从胚胎发育成成人,我们的组织生长、成熟并适应出生后环境,这些职责也会发生变化。这些其他非 ATP 相关功能与细胞用于构建、分解和回收其分子构件的大多数主要代谢途径密切相关。如果没有线粒体,细胞甚至无法制造生长和发挥功能所需的 RNA 和 DNA。RNA 和 DNA 的组成部分是嘌呤和嘧啶。
线粒体是负责氧化磷酸化的细胞器,以ATP的形式产生能量。根据主要涉及的遗传缺陷,线粒体病可分为以下几类病因(表 1):
●呼吸链蛋白
●呼吸链辅助蛋白
●线粒体RNA翻译
●线粒体内膜脂质环境
●线粒体DNA(mitochondrial DNA, mtDNA)减少
●线粒体动力学
氧化磷酸化由位于线粒体内膜的呼吸链完成。呼吸链包括5种膜内复合体和2种可移动电子载体[辅酶Q10(coenzyme Q10, CoQ10)和细胞色素c]。虽然最早报道的人类线粒体病是由线粒体DNA突变所致,但现已认识到,线粒体基因和核基因均为氧化磷酸化途径提供蛋白质。
氧化磷酸化途径的蛋白质必须得到翻译、进入线粒体并嵌入线粒体内膜。影响这些过程的基因突变可称为线粒体呼吸链受到“间接打击(indirect hits)”,也称呼吸链辅助蛋白缺陷。线粒体RNA翻译缺陷可扰乱多种氧化磷酸化过程,引起线粒体病。
线粒体病的范围已进一步扩大至包括线粒体内膜磷脂双层结构缺陷。此外,现已认识到线粒体的动力学特性;线粒体不断地移动、融合和分裂,而这些功能的缺陷构成了一类新的线粒体病。在线粒体DNA数量充足的情况下,线粒体呼吸链复合体产生能量的关键亚基才能得以维持。核基因突变所致的线粒体DNA维持缺陷会引起线粒体DNA耗竭综合征(mitochondrial DNA depletion syndrome, MDS)。由此所致的器官功能障碍可能是由于能量产生不足。
线粒体病可表现出多种临床表型,这加大了临床医生诊断该病的难度。能量需求高的组织(即脑、心、骨骼肌)优先受累。骨骼肌受累则称为线粒体肌病;脑和骨骼肌均受累则称线粒体脑肌病。
表1:与线粒体肌病相关的基因突变
| 线粒体DNA突变 |
| 编码呼吸链蛋白的基因突变 |
| 复合物 I(ND1 和 ND4 导致纯肌病;ND5 导致 MELAS/LHON/MERRF 肌病) |
| 复合物 III(细胞色素 b 导致运动不耐受/肌痛,很少导致心肌病或多系统疾病) |
| 复合物 IV(COX I、COXII、COXIII)会导致运动不耐受/肌红蛋白尿,但也可能导致严重的脑肌病(全部三种)或 MELAS(仅 COX III) |
| 影响蛋白质合成的突变 |
| 转移 RNA 突变最常导致 MELAS 或 MERRF,也可能导致孤立性肌病、CPEO 或呼吸肌无力 |
| 核糖体 RNA 突变会导致氨基糖苷类相关耳聋和心肌病,但不会导致骨骼肌病 |
| 大规模删除/重复最常导致散发性 CPEO/Kearns-Sayre 综合征,不太常见导致 Pearson 综合征 |
| 核DNA突变 |
| 编码呼吸链蛋白的基因突变 |
| 辅酶 Q10 缺乏可导致孤立性肌病或快速致命的婴儿肾病综合征脑肌病 |
| 复合体 I 突变(NDUFS1、2、3、4、6、7、8、NDUFV1、2、NDUFA1、2、11)和复合体 II 突变(SDHA)可引起常染色体隐性遗传 Leigh 综合征 |
| 复杂 II 突变(SDHB、SDHC、SDHD)可引起副神经节瘤和肉瘤 |
| 复合体 III 突变(UQCRB、UQCRQ)可导致乳酸性酸中毒、低血糖和精神运动迟缓 |
| 复杂的 IV 突变(COX6B1、COX4I2)可导致婴儿脑病、贫血和胰腺功能障碍 |
| 编码呼吸链蛋白组装或功能所需蛋白的核基因突变 |
| 复合体 I 突变(NDUFAF1、2、3、4、C20orf7、C8orf38、C6orf66)可引起多种疾病,包括 Leigh 综合征和致命性婴儿脑病 |
| 复合体 II 突变 (SDHAF1, 2) 可引起婴儿白质脑病和副神经节瘤 |
| 复合体 III 突变 (BCS1L) 可导致 GRACILE 综合征 |
| 复杂的 IV 突变(SURF 1、COX10、COX15、SCO1、SCO2)可导致 Leigh 综合征和婴儿心脑肌病 |
| 复合 V 突变(ATPAF2、ATP5A1、ATP5E、TMEM70)可引起脑病和 3-甲基戊二酸尿症 |
| 影响线粒体蛋白质合成的核基因缺陷(导致线粒体 DNA 多次缺失或耗尽) |
| POLG、C10orf2(编码闪烁解旋酶)、RRM2B、SLC25A4、POLG2 和 DGUOK 突变可导致常染色体显性或隐性 CPEO |
| TK2、SUCLA2、SUCLA2、SUCLG1、RRM2B、DGUOK突变可引起先天性脑肌病和肌营养不良样脑肌病;TK2 突变还可导致缓慢进展的全身性肌病 |
| 核基因突变导致线粒体动力学缺陷(融合/裂变) |
| OPA1 突变可引起双侧视神经病变,并可能伴有感音神经性听力损失;MFN2 突变可导致轴突形式的腓骨肌萎缩症 |
| 影响线粒体蛋白的核基因缺陷与细胞凋亡有关 |
| AIF1 突变可导致多处 mtDNA 缺失或 mtDNA 耗竭,并出现线粒体脑肌病;FASTKD2 也可能伴有线粒体脑肌病;APOPT1 突变可导致脑白质营养不良 |
| 核基因突变导致脂质环境缺陷 |
| TAZ 突变可导致 Barth 综合征、X 连锁线粒体肌病、心肌病、中性粒细胞减少症和身材矮小 |
| SERAC1突变可导致MEGDEL |
| 影响呼吸链以外线粒体代谢途径的核基因缺陷 |
| 线粒体底物转运缺陷包括CPT 1A缺乏,主要导致肝脏疾病;CPT 2 缺乏症最常导致复发性横纹肌溶解综合征 |
| 线粒体底物利用缺陷包括三功能蛋白缺乏,这会导致神经病、肌病和复发性横纹肌溶解症 |
| 核基因突变导致铁硫簇组装/稳态缺陷 |
| ISCU 缺陷与肌病、运动不耐受和复发性横纹肌溶解症相关 |
该表并未包含所有内容,而是列出了一些与人类线粒体疾病表型相关的更常见或更重要的基因。
Barth综合征:X连锁心肌病、线粒体肌病和周期性中性粒细胞减少症;CPEO:慢性进行性眼外肌麻痹;GRACILE综合征:生长迟缓、氨基酸尿、胆汁淤积、铁超负荷、乳酸性酸中毒和过早死亡;Kearns-Sayre 综合征:患有色素性视网膜病变且 20 岁之前发病的 CPEO;Leigh综合征:亚急性坏死性脑脊髓病;LHON:莱伯遗传性视神经病;MEGDEL:3-甲基戊烯酸尿症伴耳聋、脑病和 Leigh 样综合征;MELAS:线粒体脑病伴乳酸性酸中毒和中风样发作;MERRF:伴有参差不齐的红纤维的肌阵挛性癫痫;皮尔逊综合征:铁粒幼细胞性贫血和胰腺功能障碍;RNA:核糖核酸;CPT:肉毒碱棕榈酰转移酶;ISCU:铁硫簇组装体。
线粒体的多种作用线粒体含有制造血红蛋白
所需的嘧啶生物合成(二氢乳清酸脱氢酶)和血红素合成(d-氨基乙酰丙酸合成酶)的限速酶。在肝脏中,线粒体专门负责尿素循环中氨的解毒。胆固醇代谢、雌激素和睾酮合成、神经递质代谢以及自由基产生和解毒也需要线粒体。除了分解(氧化)我们吃喝的脂肪、蛋白质和碳水化合物之外,它们还完成所有这些工作。
线粒体疾病的定义
线粒体疾病是 mtDNA 或 nDNA 遗传或自发突变的结果,这些突变导致通常存在于线粒体中的蛋白质或 RNA 分子的功能改变。然而,线粒体功能问题可能只影响某些组织,这是由于发育和生长过程中发生的一些我们尚不了解的因素造成的。即使考虑线粒体蛋白的组织特异性亚型,也很难解释临床上看到的线粒体疾病综合征中受影响器官系统的可变模式。
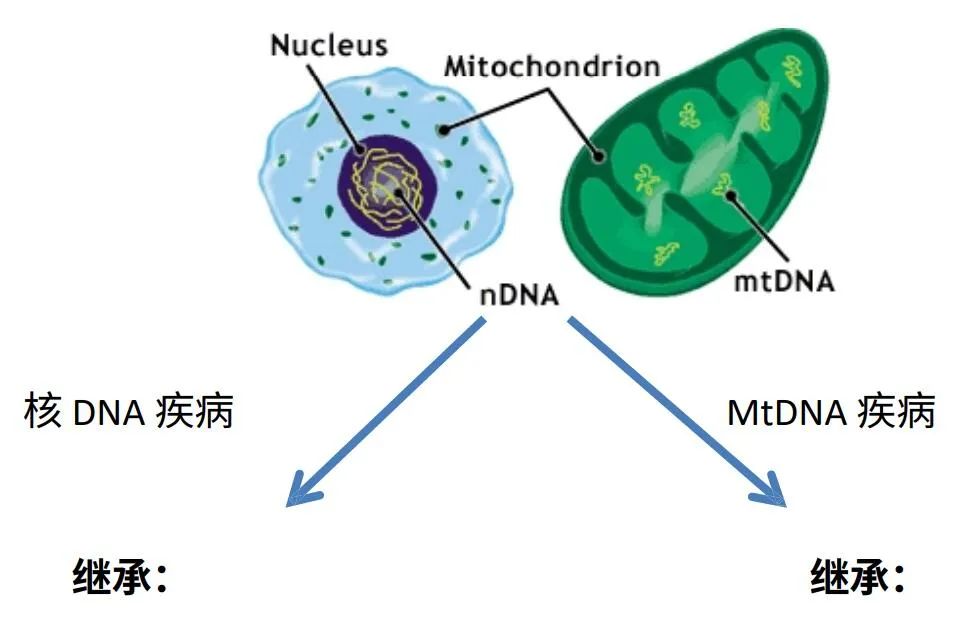
线粒体疾病的基因拷贝
由于线粒体在不同的组织中执行许多不同的功能,因此实际上有数百种不同的线粒体疾病。每种疾病都会产生一系列异常现象,这些异常现象在诊断的早期阶段可能会让患者和医生感到困惑。由于数百个基因和细胞之间复杂的相互作用,这些基因和细胞必须合作才能保持我们的代谢机制顺利运行,因此相同的 mtDNA 突变可能不会产生相同的疾病,这是线粒体疾病的一个标志。基因拷贝是由相同突变引起的疾病,但临床上可能看起来不一样。
线粒体疾病的表型
反之亦然:mtDNA 和 nDNA 的不同突变可以导致相同的疾病。在遗传学中,这些被称为表型。Leigh综合症就是一个很好的例子,它可能是由大约十几种不同的基因缺陷引起的。
Leigh 综合征最初是对一名受影响儿童大脑的神经病理学描述,由英国杰出医生 Denis Leigh 在 1951 年描述。其特征是脑干、小脑和基底神经节双侧对称 MRI 异常,并且经常出现伴有血液或脑脊液中乳酸水平升高。Leigh 综合征可能由 NARP 突变、MERRF 突变、复合物 I 缺陷、细胞色素氧化酶 (COX) 缺陷、丙酮酸脱氢酶 (PDH) 缺陷和其他未定位的 DNA 变化引起。然而,并非所有具有这些 DNA 异常的儿童都会继续患上 Leigh 综合征。
成人线粒体疾病
成人线粒体疾病更加复杂,因为随着年龄的增长,线粒体 DNA 会发生可检测到的变化,相反,衰老过程本身可能是由线粒体功能恶化造成的。成人中存在广泛的代谢、遗传和获得性疾病,其中线粒体功能异常已被假定或证实。
一、临床特征
线粒体肌病的临床表现变化多端。肌病可能是主要的起病特征,或者仅为次要特征。这些疾病轻则表现为轻度运动不耐受,重则呈致命性婴儿期脑肌病或多系统疾病。
1、线粒体肌病的临床表型可分类如下:
单纯性肌病
慢性进行性眼外肌麻痹(chronic progressive external ophthalmoplegia, CPEO)或Kearns-Sayre综合征(Kearns-Sayre syndrome, KSS)
婴儿期和儿童期脑肌病
多系统疾病伴肌病
该分类系统的局限性在于,这些表型类别之间存在一定程度的重叠。例如,CPEO患者后来可能会发生轻度近端肌无力或运动不耐受。或者,最初仅存在运动不耐受或轻度近端肌病的患者可能进展为眼外肌麻痹。
MDS这组疾病的特征是,线粒体DNA显著减少并累及多种组织,表现为肌肉受累、肝脏受累或者肌肉与脑均受累。MDS主要可分为:肌病型,TK2、RRM2B和AGK突变;脑肌病型,SUCLA2、SUCLG1和RRM2B突变;肝脑型,GDUOK、MPV17、POLG和C10ORF2突变;和/或神经胃肠型,TYMP突变。
与其他代谢性疾病一样,线粒体肌病可能在生理应激增加(例如患病或手术/麻醉)期间发病。此外,其可能与恢复延迟或横纹肌溶解发作有关。
小型研究的有限证据提示,线粒体病患者的精神病症状发生率增高,但尚缺乏大型的病例对照系列研究。线粒体病患者的精神病症状可能更难治疗,因为许多精神药物会对线粒体功能造成负面影响。例如,用作心境稳定剂的丙戊酸会抑制线粒体呼吸链,线粒体病患者应避免使用。
2、单纯性肌病
线粒体病偶尔仅表现出肌肉症状,包括运动不耐受、乏力、肌无力、血清肌酸激酶(creatine kinase, CK)升高、肌痛,较少情况下包括横纹肌溶解。肌肉受累情况可能有很大差异。肢体近端肌肉更易受累,但远端肌病也有报道。
单纯性肌病的遗传缺陷可能是线粒体或核基因突变:
线粒体DNA突变所致的单纯性肌病较罕见,文献中仅报道了几个病例
单纯性肌病可能是线粒体DNA突变所致数种多系统线粒体病的初始表现
许多核DNA缺陷可表现出肌病的症状体征(表 1)。这些缺陷包括呼吸链缺陷、辅助蛋白缺陷、基因组间信号传导缺陷和线粒体内膜脂质环境缺陷。
核DNA突变所致辅酶Q10缺乏可表现为孤立的近端肌无力,但其他临床表现包括脑肌病、小脑性共济失调、肾病综合征和多系统疾病。识别出这种疾病很重要,因为补充辅酶Q10也许能治疗该病。
少数TK2基因突变所致DNA减少的患者会发生缓慢进展性全身性肌病,主要累及中轴肌和近端肌肉,但呼吸、面部和眼部肌肉也会受累。此外,在婴儿期和儿童期严重线粒体DNA丢失患者中,已报道了TK2基因突变所致线粒体DNA丢失呈以肌病为主的快速进展性表现。
3、CPEO和Kearns-Sayre综合征
CPEO患者通常会在30-40岁发生缓慢进展性眼外肌轻瘫伴双侧上睑下垂,但这些表现也可在任意年龄出现。患者通常没有复视或仅有一过性复视。CPEO患者通常存在轻度早期残疾,但详细评估视觉常会发现显著的视觉缺陷。
CPEO的鉴别诊断包括重症肌无力、眼部肌炎、甲状腺相关眼眶病、眼咽型肌营养不良和先天性眼外肌纤维化。
KSS是指CPEO合并色素性视网膜病,20岁前发病。也可能存在其他异常表现,包括身材矮小、小脑性共济失调、脑脊液(cerebrospinal fluid, CSF)蛋白增加(>100mg/dL)、心脏传导缺陷、贫血、糖尿病、耳聋,以及认知缺陷/智力障碍。KSS通常比单纯性CPEO更严重,将进展成完全眼肌麻痹,患者常因相关缺陷在三十多岁前死亡。CPEO或KSS患者均可出现近端肌病。
CPEO和KSS可能是散发性、母系遗传、常染色体显性遗传或常染色体隐性遗传,表明线粒体和核DNA缺陷都可引起相同的表型。当缺陷表现是线粒体DNA缺陷所致时,通常原因是大规模线粒体DNA重排,但也有点突变的罕见报道。
常染色体显性和隐性遗传性CPEO与多个核DNA基因突变有关,包括POLG、C10orf2、RRM2B、SLC25A4、POLG2、DGUOK和SPG7,它们会引起基因组间信号传导缺陷,导致多重继发性线粒体DNA缺失。
POLG的致病突变与两种表型有关:不伴全身受累的常染色体隐性遗传性进行性眼外肌麻痹(autosomal recessive progressive external ophthalmoplegia, arPEO),以及伴全身受累的常染色体显性遗传性进行性眼外肌麻痹(autosomal dominant progressive external ophthalmoplegia, adPEO),其中全身受累包括大多数患者都存在的全身性肌病以及不同程度的感音神经性聋、轴索型神经病、共济失调、抑郁、帕金森综合征、性腺功能减退症及白内障,这种疾病也称CPEO+。
4、Leber遗传性视神经病
LHON属于母系遗传的双侧亚急性视神经病变。LHON年轻男性患者通常会出现严重的永久性视力丧失。除了2种罕见的独特线粒体DNA突变之外,LHON通常不伴肌病,但一些患者会表现出运动障碍或心脏传导缺陷。
5、婴儿期或儿童期严重脑肌病
婴儿期与儿童期早期出现的线粒体病具有多种不同表现,但常见肌病。
一项回顾性研究纳入1975-2006年间的32例原发性线粒体病新生儿,发现脑肌病型(脑病、癫痫发作、肌张力减退和/或眼病的孤立性或组合性症状体征)是最常见的线粒体病表现,见于其中17例患者(53%)。其他表现包括:肝肠型(25%);心脏型(16%),主要表现为早期致命性心肌病;Barth综合征(6%),即X连锁心肌病、线粒体肌病和周期性中性粒细胞减少。患者预后不佳。总体上,13例患者在新生儿期死亡,另15例在稍后死亡,2例失访,2例患者存活。
最严重形式的婴儿期或儿童期严重脑肌病在出生时表现为明显肌张力减退、需要通气支持的呼吸肌无力以及喂养困难。受累婴儿一般在1岁前死亡,所以称为“致命性婴儿期线粒体病”。患儿可能伴有脑、心、肝或肾受累。原因通常是TK2或SUCLA2基因突变所致MDS。一些儿童伴有广泛性近端肾小管功能障碍(即Fanconi综合征)所致的严重肾衰竭,且有复合物Ⅲ缺陷。辅酶Q10缺乏也可导致快速致命的婴儿期脑肌病伴肾病综合征。
TK2或SUCLA2基因突变也可引起类似于肌营养不良的儿童期起病表型。血清肌酸激酶水平持续升高以及类似于Duchenne型肌营养不良的膜异常使得该病的鉴别更加复杂。
有患者表现为婴儿期出现严重单纯性肌病,无多系统受累。一小部分的COX亚基缺乏患儿在随后2年症状自发改善,通常到2岁或3岁时恢复正常。这种罕见疾病的分子基础是线粒体DNA tRNA突变。
POLG相关疾病可在婴儿期或成人期引起严重脑病。例如,儿童期肌脑肝病谱系,发病时间为3岁前,特征为发育延迟、乳酸酸中毒、肌病和生长迟滞。Alpers-Huttenlocher综合征也是POLG相关疾病,属于严重表型,特征为儿童期发病的进行性脑病、难治性癫痫及肝衰竭。
6、多系统受累为主的疾病伴肌病
多系统受累是线粒体病的标志性特点之一。多系统受累为主的疾病患者会出现以下表现的不同组合:中枢和/或周围神经系统受累,眼科异常,感音神经性耳聋,胃肠道症状,心、肝、肾疾病,内分泌功能障碍,以及生长障碍(身材矮小)。患者通常严重残疾。
表现为多器官系统受累的公认临床综合征的线粒体病包括以下几类:
Barth综合征(X连锁心肌病、线粒体肌病和周期性中性粒细胞减少)
生长迟滞、氨基酸尿症、胆汁淤积、铁过载、乳酸酸中毒和早期死亡(growth retardation, amino aciduria, cholestasis, iron overload, lactic acidosis, and early death, GRACILE)
LHON
Leigh综合征(亚急性坏死性脑脊髓病)
母系遗传性耳聋和糖尿病(maternally inherited deafness and diabetes, MIDD)
线粒体脑肌病伴乳酸酸中毒和脑卒中样发作(mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes, MELAS)
线粒体神经胃肠型脑病(mitochondrial neurogastrointestinal encephalopathy, MNGIE)
肌阵挛性癫痫伴碎红纤维(myoclonic epilepsy with ragged red fibers, MERRF)
神经病变、共济失调和视网膜色素变性(neuropathy, ataxia, and retinitis pigmentosa, NARP)
Pearson综合症(铁粒幼细胞贫血与胰腺功能障碍)
其中,Leigh综合征和MELAS最常见。遗传异常根据综合征不同而异。
很大一部分有上述多系统疾病的患者很可能存在从未被注意到或诊断出的显著肌肉受累,这可能是因为疾病迅速致命或者严重残疾妨碍了对肌病的识别。
MELAS、MERRF、MNGIE及Leigh综合征患者在详细评估时可表现出显著的肌病改变。然而,这些病症中的肌病通常不会导致功能显著受损。
例如,一项研究评估了50例MELAS患者,其均存在tRNA m.3243A>G突变,而80%的MELAS都是由该突变引起。仅基于临床,一半患者存在肌病证据,多达72%的活检标本显示符合线粒体肌病的组织学异常。中度肢体无力是最常见的肌病体征。患者可能存在上睑下垂和眼外肌麻痹。
7、Leigh综合征
Leigh综合征(亚急性坏死性脑脊髓病)通常在婴儿期或儿童期早期发病,但也有儿童期后期和成人期发病的报道。该病特点为发育延迟或精神运动退化、共济失调、肌张力障碍、眼外肌麻痹、癫痫发作、乳酸酸中毒、呕吐和无力。一些患者会出现肌病和周围神经病变。当出现非典型和/或非神经系统特征时,例如糖尿病、身材矮小、多毛症、心肌病、贫血、肾衰竭、呕吐或腹泻,称为“Leigh样综合征”。
Leigh综合征的病理学标志是基底节、丘脑、脑干和脊髓出现双侧对称性坏死性病变伴海绵状改变和微囊。乳头体不受累。脑MRI的T2和液体衰减反转恢复序列图像示壳核、基底节和脑干出现异常白质信号。
患者预后通常不良,发病后一般仅存活数月。
Leigh综合征表型由多种机制引起的线粒体代谢变化导致,包括丙酮酸脱氢酶复合物异常和由核DNA或线粒体DNA突变引起的呼吸链功能障碍。该综合征具有遗传异质性,在超过85个基因中识别出了致病突变。
8、MELAS
MELAS综合征为线粒体DNA突变引起的母系遗传性多系统疾病。此综合征的特征为出现脑卒中样发作,导致轻偏瘫、偏盲或皮质盲。其他常见特征包括局灶性或全面性癫痫发作、复发性偏头痛样头痛、呕吐、身材矮小、听力损失以及肌无力。许多tRNA突变都可引发MELAS,但80%的病例与m.3243A>G突变有关,10%与m.3271T>C tRNA突变有关。
MELAS患者出现的脑卒中样发作特征为神经系统症状急性发作,以及脑弥散加权MRI显示高信号。出于以下几种原因,这些发作不同于典型栓塞性或血栓形成性缺血性脑卒中,因此称为“脑卒中样发作”:
脑部病灶并不遵循血管分布。
MRI表观弥散系数不一定降低(组织梗死时会降低),甚至可能升高或出现混合模式。
急性MRI信号改变并不静止不变,可能移行、波动或消退,并且变化比典型缺血性脑卒中更迅速、更常见。
MELAS通常在正常早期发育之后的儿童期显现。复发-缓解病程最常见,伴脑卒中样发作,导致进展性神经功能障碍和痴呆。
MELAS最初的诊断标准要求在40岁前出现卒中样发作,以癫痫发作或痴呆为特征的脑病,以及血液乳酸酸中毒或骨骼肌活检显示碎红纤维。然而,目前认识到更广泛的表型也符合该诊断,其中包括40岁以后临床发病。
POLG突变也与儿童期或成人期脑卒中样发作伴枕叶明显受累有关。发生脑卒中的患儿常见癫痫发作和肝功能障碍或衰竭。
9、MERRF MERRF的特征为肌阵挛(通常为首发症状),伴全面性癫痫、共济失调和肌病。其他特征包括痴呆、视神经萎缩、双侧耳聋、周围神经病变、痉挛状态、脂肪过多症和/或心肌病伴Wolff-Parkinson-White综合征。通常在正常早期发育之后的儿童期发病。
MERRF由线粒体DNA突变引起。在超过80%的MERRF患者中,编码tRNA(赖氨酸)的线粒体MT-TK基因存在突变,即核苷酸8344位点的A>G突变。
10、MEMSA
和MERRF相似,肌阵挛性癫痫、肌病及感觉性共济失调(myoclonic epilepsy, myopathy, and sensory ataxia, MEMSA)综合征是由POLG致病突变引起的一系列疾病,表现为癫痫、肌病及共济失调但不伴眼肌麻痹。
11、MIDD
尽管此表型表现多样,但MIDD的特征皆有胰岛素分泌缺陷(会发展成胰岛素依赖)和感音神经性(耳蜗性)耳聋。糖尿病与听力损失的平均发病年龄为30-40岁。MIDD常伴发的其他异常包括黄斑视网膜营养不良、肌病、心脏疾病、妊娠期糖尿病、肾脏疾病(尤其是局灶节段性肾小球硬化)、身材矮小和胃肠道疾病。
一项研究纳入了51例被诊断为MIDD患者的数据,发现22例(43%)患者存在肌病。肌病通常累及肢体近端,并且可能伴有运动诱发性痛性痉挛和无力。
MIDD由线粒体DNA tRNA基因核苷酸3243位点的A-G突变所致,80%的MELAS病例也由同一突变引起。部分患者的表现有重叠,可见A3243G突变可引起多种表现,既可为仅有糖尿病或听力损失,也可为MIDD,甚至是MELAS。
12、MNGIE
MNGIE为一种多系统线粒体病,其特征为进行性严重胃肠动力障碍和恶病质,上睑下垂、不伴复视的眼肌麻痹或眼肌瘫痪,对称性多发性神经病,以及无症状性白质脑病。
MNGIE的胃肠动力障碍与假性梗阻可能由内脏线粒体肌病引起。常见症状包括早饱、恶心、吞咽困难、胃食管反流、餐后呕吐、发作性腹痛、腹部膨隆和腹泻。神经病变以脱髓鞘为主,但也常同时发生轴突受累。症状包括感觉异常、疼痛和远端无力。
MNGIE的发病年龄、症状出现顺序以及疾病进展速度均有很大差异性。发病年龄介于几岁至四十几岁,但60%-73%的患者在20岁之前发病。
MNGIE的长期预后不良。两项分别纳入35例和102例患者的研究显示,平均死亡年龄分别为35岁和38岁(范围为15-58岁)。
MNGIE是一种核DNA疾病。大多数病例是由编码胸苷磷酸化酶的TYMP基因(也称ECGF1基因)突变导致,该突变会引起线粒体DNA继发性减少和/或多重缺失。该病为常染色体隐性遗传。在一项病例报告中,1例成人患者有类似于MNGIE的表型但血浆胸苷水平正常,结果发现其RRM2B基因中有2个致病突变。随后一项报道通过31例存在RRM2B致病突变的患者发现(包括5例通过回顾文献挑选的患者),19%的患者明显存在胃肠道症撞。
13、NARP
NARP的特征为发育延迟、感觉性多发性神经病、共济失调、色素性视网膜病、肌无力、癫痫和痴呆的不同组合。该病最常在儿童晚期或成人期发病。
NARP是由MT-ATP6基因的核苷酸8993位点T>G突变引起。该基因还与母系遗传的Leigh综合征相关。一些证据表明,与NARP表型相比,Leigh综合征表型与更高的T8993G组织突变负荷有关。
14、Pearson综合征
Pearson综合征(铁粒幼细胞贫血与胰腺功能障碍)是一种先天性多系统疾病,其特征为严重贫血、骨髓中有环形铁粒幼细胞、中性粒细胞减少、血小板减少以及胰腺外分泌功能不全。该病由线粒体DNA缺失(缺失大小为2-10kb)引起。血液中线粒体DNA缺失的比例通常比其他组织中更高。
Pearson综合征通常在婴儿期致命。存活超过婴儿期的患者会出现KSS的症状体征。
15、辅酶Q10缺乏
有辅酶Q10生物合成障碍的患者会发生原发性辅酶Q10缺乏。继发性辅酶Q10缺乏是指线粒体呼吸链疾病导致辅酶Q10水平降低,但其原因不是辅酶Q10生物合成途径的原发性缺陷。
辅酶Q10的几种作用包括:作为线粒体呼吸链中的电子载体,作为脂溶性抗氧化剂,参与DNA复制和修复所需的嘧啶合成,以及参与细胞膜调节。
现已报道了辅酶Q10缺乏的5种主要表型,包括:
小脑性共济失调
婴儿期重度多系统疾病
肾病
单纯性肌病
脑肌病
然而,需要注意的是,辅酶Q10缺乏症具有临床异质性并且存在其他表型。
二、评估与诊断
由于累及线粒体功能的病症可来源于两种基因组(核基因和线粒体基因)、具有多系统表现和广泛的表型异质性,所以通常难以诊断这些疾病。患者在首次就医时,临床表型的体现常常并不完整,这进一步加大了诊断难度。评估和诊断方法根据患者年龄、临床表型和推定遗传模式的不同而有差异。
对于有某种母系遗传综合征(如LHON、MELAS、MERRF、MIDD或NARP)典型表型的患者,初始检查应包括适当的线粒体DNA检测。若基因检查可确认疾病类型,则此方法可省去肌肉活检或详尽的代谢评估。
同样地,对于表现出核DNA疾病(已确定某个基因或相关基因联系,如常染色体CPEO或MNGIE)典型特征的患者,初始检查应包括分子遗传检查,以确认是否有致病性核突变。
若成人出现提示线粒体病的非特异性临床表现,应根据完整病史和体格检查结果来选择检查内容,并首先进行基本的实验室检查。根据主诉症状和体征,可能需要进行额外检查来排除鉴别诊断中的其他疾病
大多数情况下,应先进行基因检测,后进行侵入性检查(如肌肉活检)。
若基因检测不能确诊或需要排除鉴别诊断中的其他疾病,则建议行肌肉活检。
对于出现复杂的神经系统或多系统受累的儿童,通常需进行下述全面线粒体评估。
1、病史和体格检查
包括至少3代人信息的详尽家族史很重要。特别是,应确定家族是否有新生儿期或儿童期死亡,以及任何家族成员是否存在多系统疾病特征的病史。
若有母系遗传史,即仅由女性遗传疾病且未观察到男性-男性遗传,则可怀疑为线粒体病。对于异质性突变,若突变负荷低于引起症状所需的阈值,母系遗传现象可能不明显。而且,线粒体DNA突变可能为散发性,这些患者的其他家庭成员不受累。另外,据估计,约有1700种线粒体蛋白由核DNA编码。线粒体DNA仅编码13种多肽、2种核糖体RNA和22种tRNA。因此可以预料,更大一部分线粒体功能障碍与核DNA有关,并且是常染色体显性或隐性遗传模式。
线粒体病的一个重要线索为多系统受累病史,尤其是在最易发生线粒体缺陷的器官中。提示多系统受累的病史和体格检查表现包括:
脑–脑卒中样发作、癫痫发作、肌阵挛、共济失调、发育迟缓或退化、痴呆、偏头痛和肌张力障碍
眼–色素性视网膜病、视神经萎缩和白内障
骨骼肌–运动不耐受、肌痛、横纹肌溶解或肌红蛋白尿、肌无力(主要累及近端)、肌张力减退、上睑下垂以及眼外肌运动障碍伴或不伴复视
神经病变和自主神经功能障碍
心脏–心脏传导缺陷和心肌病
内分泌–糖尿病和甲状旁腺功能减退
肾–近端肾单位功能障碍和肾小球病
胃肠道–动力改变、肝病、恶心和呕吐发作以及胰腺外分泌功能障碍
皮肤–多发性脂肪瘤
血液系统–铁粒幼细胞贫血和全血细胞减少
代谢性酸中毒
身材矮小
其他相关发现包括同一患者具有神经病变与肌病的病史。
2、实验室检测
全血细胞计数、血清肌酸激酶和尿酸、血清转氨酶、血清白蛋白、血清乳酸和丙酮酸、若血清乳酸升高的话,检测乳酸/丙酮酸比值、血清氨基酸类(检查丙氨酸是否升高)、血清酰基肉碱(游离肉碱水平低和酰基/游离肉碱比值升高提示脂肪酸氧化受损)、血清和尿3-甲基戊烯二酸、尿有机酸定量或定性检查(检查三羧酸循环中间体、甲基丙二酸和二羧酸是否升高)、采集脑脊液后应检测乳酸、丙酮酸、氨基酸类和5-甲基四氢叶酸(脑叶酸缺乏时会出现5-甲基四氢叶酸异常,数种线粒体病可出现脑叶酸缺乏)
存在CNS症状时,例如脑肌病儿科患者,脑脊液分析可能有帮助。但是,若患者没有CNS症状,例如单纯性肌病或CPEO患者,脑脊液分析不太可能有用。
血清肌酸激酶水平通常正常或仅轻微升高,但在严重肌病患者中,血浆肌酸激酶水平正常可能是线粒体病的征象。
当糖酵解速度超过丙酮酸的利用时,即可发生乳酸升高。据报道,乳酸水平升高的特异性为34%-62%,敏感性为83%-100%,但还应考虑到一些因素:
一些技术因素可使乳酸和丙酮酸假性升高。例如,样本未用冰块恰当运输或采血时使用了止血带,或⼉童在采样过程中挣扎、哭闹都会导致乳酸假性升高。
乳酸升高并非线粒体病的特异性表现,先天性代谢病、毒素暴露、组织缺血和其他疾病(如硫胺素缺乏)也可出现乳酸升高。
餐后乳酸水平比空腹乳酸水平更具敏感性。血清和脑脊液乳酸/丙酮酸比值仅在乳酸水平升高时有帮助。
某些线粒体病(如LHON、Leigh综合征、KSS和POLG相关线粒体病)的乳酸水平至多轻微升高。其他线粒体病患者可能仅在生理应激期间才会出现乳酸水平升高。
⼤多数诊断算法建议评估⾎液、尿液和脊髓液中选定的线粒体⽣物标志物,这些指标通常包括⾎浆和脑脊液 (CSF)中的乳酸、丙酮酸、血氨基酸肉碱、尿氨基酸肉碱与有机酸。
在正确收集的样本中,⾎浆乳酸显着升⾼(>3 mmol/ l),表明存在线粒体功能障碍,这可能是由于原发性线粒体疾病引起的,也有可能是有机酸⾎症、其他先天性代谢缺陷、毒素、组织缺⾎和某些其他疾病引起(其他疾病继发性引起)。
多项研究表明,在原发性线粒体疾病患者中,真正升⾼的乳酸⽔平的敏感性为 34% ⾄ 62%之间,特异性为 83% ⾄ 100 %之间。⾎乳酸/丙酮酸⽐率对于区分电⼦传递链 (ETC) 疾病与丙酮酸代谢紊乱最为可靠,但只有当乳酸水平升高时才会出现这种情况。
脑脊液乳酸升⾼,可能是具有相关神经系统症状的患者,确诊线粒体疾病的有⽤标志。虽然各种脑部疾病,特别是癫痫持续状态,可以暂时增加脑脊液乳酸, 但收集伪影问题不大。令⼈惊讶的是,尿乳酸与线粒体疾病的存在相关性不是很明确。
丙酮酸升⾼是一种有用的生物标志,可用于检测与丙酮酸代谢密切相关的酶,特别是丙酮酸脱氢酶和丙酮酸羧化酶缺陷。⾎液丙酮酸⽔平也受到收集和处理错误的困扰;此外,丙酮酸是⼀种⾮常不稳定的化合物。
在评估可能患有线粒体疾病的患者时,通常需要对⾎液或脊髓液中的氨基酸进⾏定量分析。⼏种氨基酸发⽣升⾼由于呼吸链功能障碍(包括丙氨酸、⽢氨酸、脯氨酸和苏氨酸)导致氧化还原状态改变。原发性线粒体疾病患者中丙氨酸或其他氨基酸升⾼的确切敏感性和特异性尚不清楚。⾎液或脊髓液中可能存在升⾼,并且显着的发现可能仅在临 床恶化时出现。尿液氨基酸最常⽤于评估线粒体疾病相关的肾⼩管病。
⾁碱充当游离脂肪酸的线粒体穿梭器,也是具有潜在毒性的辅酶 A 酯的关键受体。肉碱可以恢复线粒体内辅酶 A, 并去除酯化中间体。通过对⾎液总⾁碱和游离⾁碱⽔平的定量分析,以及酰基⾁碱分析,可以识别原发性或继发性脂肪酸氧化缺陷,以及⼀ 些原发性氨基酸和有机酸⾎症。尽管各种线粒体综述中都建议进⾏酰基⾁碱检测,但明确⽀持这⼀建议的背景⽂献有限。之所以建议进⾏此项测试,因为线粒体疾病患者可能存在继发性脂肪酸氧化紊乱,并且某些线粒体表型与其他先天性代谢缺陷重叠,而酰基⾁碱分析可对其进行诊断。
线粒体疾病患者的尿液有机酸经常出现变化。对 67 名线粒体疾病患者和 21 名有机酸⾎症患者的样本进⾏回顾性分析,发现苹果酸和富⻢酸的升⾼与线粒体疾病最相关。其他柠檬酸循环中间体和乳酸相关性较差。某些原发性线粒体疾病,可出现轻度⾄中度的3- 甲基戊⼆酸 (3MG) 升⾼、⼆羧酸尿症、2-氧代⼰⼆酸尿症、2-氨基⼰⼆酸尿症和 甲基丙⼆酸尿症。虽然尿液有机酸可以检测3MG 升⾼,但⾎液和尿液中3MG的特异性定量更可靠,特别是当 MG⽔平不显着升⾼时。
肌酸磷酸激酶和尿酸升⾼,常见于脂肪酸氧化紊乱患者的急性横纹肌溶解症中,其升⾼是由核酸和核苷酸分解代谢引起的。虽然对原发性线粒体疾病中没有进⾏⼴泛研究,但原发性线粒体疾病患者疾病可能会出现肌⾁疾病 (尤其是细胞⾊素 B 疾病和胸苷激酶 2 缺乏症) ,而原发性或继发性脂肪酸氧化障碍也可能导致,肌酸磷酸激酶和尿酸升⾼。⾎液学异常可以通过全⾎细胞计数来检测。据报道,⼀些原发性线粒体疾病会导致再⽣障碍性贫⾎、巨幼细胞性贫⾎和红细胞增多症、⽩细胞减少症、⾎⼩板减少症和全⾎细胞减少症。
多种原发性线粒体疾病与基于mtDNA 耗竭和/或⼀般肝功能障碍的肝脏病理学相关,转氨酶和⽩蛋⽩⽔平可能有助于诊断。线粒体疾病新生物指标物:FGF21 和还原型⾕胱⽢肽等有待验证。
脑叶酸缺乏常⻅于多种神经系统和代谢疾病,包括线粒体疾病,可通过测量脑脊液中的 5-甲基四氢叶酸来诊断。脑叶酸缺乏最初是在卡恩斯-赛尔综合征 (KSS) 患者的线粒体疾病中被发现的。最近对 KSS 患者的病例系列进⼀步证实了这⼀发现。在mtDNA缺失、POLG 病、和经⽣化诊断的复合物 I 缺乏症的患者中也发现脑叶酸缺乏症。
检测血液、尿液、脑脊液的共识建议:
1. ⾎液中线粒体疾病的初步评估应包括全⾎细胞计数、肌酸磷酸激酶、转氨酶、⽩蛋⽩、乳酸和丙酮酸、氨基酸和酰基⾁碱,以及尿有机酸的定量和定性。必须⼩⼼确保样本采集正确,特别是对于乳酸和丙酮酸测量。
2. 餐后乳酸⽔平⽐空腹样本更敏感,所以尽可能⾸选空腹乳酸样本。必须注意不要过度解释餐后乳酸的⼩幅升⾼。
3.只有当酸水平升高时,血液或者脑脊液里的乳酸/丙酮酸比值才有价值。
4. 对于接受线粒体疾病评估的患者,除了尿液有机酸外,还应尽可能对⾎浆和尿液中的 3- 甲基戊⼆酸(3MG)进⾏定量测定。
5. 对于怀疑患有线粒体疾病的有肌⾁症状的患者,应评估肌酸磷酸激酶和尿酸。
6.评估线粒体⼩管病时应进⾏尿液氨基酸分析
7. 获得脑脊液后,应送去进⾏乳酸、丙酮酸、氨基酸和5-甲基四氢叶酸测量。
8. 需要对其他⽣物标志物(如 FGF21、⾕胱⽢肽和脑脊液新蝶呤)进⾏进⼀步研究。
3、其他检查
空腹血糖和糖化血红蛋白、肾功能检查、心电图、神经影像学检查、肌电图(electromyography, EMG)、肌肉活检、超声心动图、眼科检查、听力检查、甲状腺和甲状旁腺检查、对脑病或癫痫发作患者进行脑电图检查、对主要表现为运动不耐受的患者进行运动试验
4、DNA检测
原发性线粒体疾病是由⺟系遗传的 mtDNA 或许多nDNA基因中的一个基因的突变引 起的。mtDNA基因组测序和异质性分析现在可以在⾎液中有效地进⾏,但可能需要测试受影响器官中的其他组织。更新的测试⽅法可以更准确地检测⾎液中低⾄ 5-10%和1-2% 的异质性。总体⽽⾔,依赖于⼤规模旁路或下⼀代测序的新技术的出现(NGS) ,已成为 mtDNA 基因组测序的新⾦标准⽅法,因为它们可以显着提⾼ mtDNA 基因组分析的点突变、低⽔平异质性和缺失的可靠性和灵敏度,从⽽提供单⼀测试来准确诊断 mtDNA 紊乱。
这种新⽅法可被视为线粒体基因组综合分析的⼀线测试,对⾎液、尿液或组织中线粒体基因组进行全面分析,确定致病性线粒体疾病突变可以让患者家庭结束诊断之旅,并接受适当的遗传咨询、携带者检测和选择性产前诊断。
作为对疑似线粒体疾病患者的诊断评估的⼀部分,可能有必要优先测试其他组织。
鉴于肾上⽪细胞中 mtDNA 含量⾼,尿液越来越被认为是 mtDNA 基因组分析的有⽤样本。这⼀发现特别适⽤于 MELAS(线粒体脑肌病、乳酸性酸中毒和中⻛样发作)综合征及其最常⻅的疾病。MTTL1 中常⻅突变 m.3243 A>G,⻣骼肌或肝脏是mtDNA 基因组测序的⾸选组织来源,因为它们的mtDNA含量⾼,依赖线粒体呼吸,并且它们可能蕴含血液里不存在的组织特异性mtDNA 突变。
mtDNA 缺失和重复综合征表现为三种表型:KSS、慢性进⾏性外眼肌⿇痹和皮尔逊综合征。检测mtDNA 缺失最常见的⽅法包括 Southern 印迹分析和⻓程(缺失特异性)聚合酶链反应分析。然⽽,Southern印迹分析缺乏⾜够的灵敏度来检测低⽔平的异质性缺失。相⽐之下,阵列⽐较基因组杂交可检测缺失,并估计缺失断点和缺失畸变率。所有这些⽅法都正被线粒体全基因组的 NGS 所取代,NGS对整个mtDNA基因组的均匀覆盖足够深,可灵敏地检测单个或多个缺失并确定其特征。
mtDNA缺失综合征是⼀组具有遗传和临床异质性的疾病,其特征是受影响组织中的mtDNA拷⻉数显着减少。mtDNA⽣物发⽣或维持异常是此类线粒体疾病的病理⽣理学基础。它们通常是由于在线粒体脱氧核苷酸合成或mtDNA复制过程中nDNA基因突变引起的。因此,诊断需要对受影响组织中mtDNA进⾏定量,通常是在正常年龄、性别和组织特异性对照的平均值以下,并与nDNA组织含量进行归一化,确定显著下降的mtDNA含量。
有1,400 多个核基因直接或间接参与线粒体功能。除了单基因测试之外,还有许多诊断实验室提供基于下⼀代多基因测序的⾯板。⼀些公司提供含有少量⽬标基因的组合,每种线粒体疾病表型(例如, 线粒体耗竭综合征)有⼏个到⼗⼏个左右。还提供超过 100、400 或 1,000 个核基因的更⼤⾯板。全外显⼦组 测序于 2011 年进⼊临床,它是⼀种越来越常⻅的诊断⼯具,⽤于疑似线粒体疾病的患者。许多研究报告描述了通过全外显⼦组测序检测核线粒体基因中的新型致病性突变,但尚未就单基因测序、 核基因组或全外显⼦组的使⽤建⽴明确的基于证据的实践建议。
DNA 检测的共识建议
1. mtDNA 基因组的⼤规模并⾏测序/NGS 是检测 mtDNA 时 的⾸选⽅法,并且应在疑似线粒体疾病的情况下进⾏,⽽不是检测有限数量的致病点突变。
2. 由于 mtDNA 突变且⾎液检测呈阴性⽽极有可能患有线粒体疾病的患者,应在其他组织中进⾏ mtDNA 评估,以避免组织特异性突变缺失或⾎液异质性⽔平较低的可能性;基于组织的检测还有助于评估其他器官受累的⻛险和家庭成员的异质性,并指导遗传咨询。
3. 尿液中的异质性分析可以选择性地⽐单独的⾎液检测提供更多信息,也更准确,特别是在常⻅的 m.3243 A>G突变而导致MELAS病例中。
4、对于疑似线粒体疾病的病例,特别是在所有接受组织活检诊断的患者中。应通过NGS对mtDNA基因组进行缺失与重复检测
如果使⽤基于聚合酶链反应的分析鉴定出单个⼩缺失,则应谨慎将这些发现与原发性线粒体疾病联系起来。当发现多个 mtDNA 缺失时,建议对参与 mtDNA⽣物合成的核基因进⾏测序。
5. 当获取组织样本⽤于线粒体研究时,应强烈考虑通过实时定量聚合酶链反应检测 mtDNA 含量(拷⻉数)进⾏ mtDNA 消耗分析,因为在⾎液中可能⽆法检测到 mtDNA 消耗。
线粒体DNA增殖是⼀种⾮特异性代偿性发现,可⻅于原发性线粒体疾病、继发性线粒体功能障碍、肌病、肌张⼒低下,并且是定期剧烈运动的副产品。
6. 当考虑对可能患有原发性线粒体疾病的患者进⾏核基因检测时,⾸选能够完全覆盖已知线粒体疾病基因的 NGS ⽅法。通常应避免单基因测试,因为不同基因的突变可以产⽣相同的表型。如果通过已知的 NGS 基因组未识别出已知突变,则应考虑全外显⼦组测序。
5、组织病理学和⽣化检测
病理学组织活检(通常是肌⾁)通常被认为是线粒体诊断的⾦标准, 尽管该测试受到敏感性和特异性有限的影响。组织活检通常用于各种组织学、⽣化和遗传学研究。随着新的分⼦检测的出现,不再需要主要依靠组织的⽣化检测来进⾏诊断,但选择性检测组织仍然是⼀种信息丰富的程序,特别是对于线粒体疾病等临床异质性疾病。组织检测可以检测具有组织特异性或低⽔平异质性的 mtDNA 突变,并 对 mtDNA 含量(拷⻉数)进⾏定量。对于肌病患者,可以通过肌⾁活检排除某些其他神经肌⾁疾病。
关于患者是否需要进⾏开放性活检以保留组织学并进⾏所有必要的测试存在争议。⼀些中⼼在进⾏线粒体针刺活检⽅⾯经验丰富。开放线粒体组织活检需要与外科医⽣可能执⾏的常规活检不同的技术。
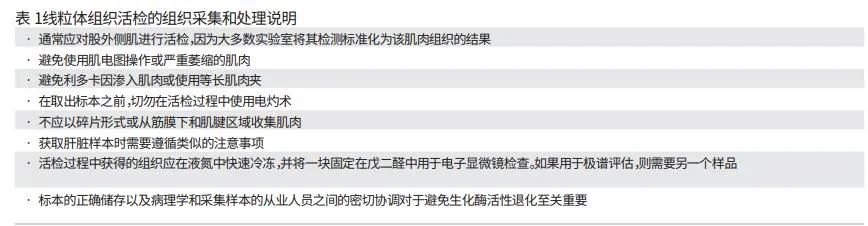
肌⾁组织常规检测包括苏⽊精和伊红 (H&E)、Gomori 三⾊染色法(⽤于不规则的粗红⾊纤维)、SDH(⽤于富含 SDH 或不规则的蓝⾊纤维)、NADH-TR(NADH-四唑还原酶)、COX(⽤于 COX 阴性纤 维)、以及联合 SDH/COX 染⾊(特别适⽤于 COX 中间纤维)。应常规使⽤机构病理科的其他标准染⾊来探索 可以诊断的其他肌病(即糖原、脂质染⾊)。电⼦显微镜 (EM) 检查线粒体内含物和超微结构异常。⼉科患者出现组织病理学异常的可能性较⼩,尽管也可以看到正常结果,但可能仅在肌⾁电镜上注意到不规则性。
线粒体疾病引起的肝功能障碍最常⻅于⼉科患者。肝活检可以显⽰线粒体肝病的选择性组织学和超微结构特征,例如脂肪变性、胆汁淤积、结构破坏以及嵴肿胀的⾮典型线粒体造成的细胞质拥挤。对于不明原因的胆汁淤积,尤其是伴有脂肪变性和肝细胞嗜酸粒细胞增多症时,应常规进⾏超微结构评估。
病理学检测的共识建议
当 DNA 检测⽆法确诊时,应在线粒体疾病的常规分析中进⾏肌⾁(和/或肝脏)活检。
2. 进⾏肌⾁活检时,在线粒体疾病的常规分析中,⾸选开放式活检,除⾮进⾏活检的中⼼具有通过经⽪活检获得⾜够质量和数量的组织的经验。
3. 股外侧肌是评估线粒体疾病时肌⾁活检的⾸选部位,因为⼤多数实验室已使⽤该部位来建⽴参考范围。
4. 在线粒体疾病组织的常规分析中应获得 COX、SDH、NADHTR 以及 SDH/COX 联合染⾊和 EM;强烈建议接受组织活检的⼉科患者进⾏ EM,因为组织学结果通常有限。
5. 线粒体肝病可能具有特征性的发现肝活检组织学⽅⾯的研究。
6. 如果可能,应冷冻多余的组织以进⾏额外的测试 。
组织⽣化测试。组织(通常是肌⾁)的功能性体外测定⼀直是线粒体疾病诊断的⽀柱,特别是在基因组学取得最新进展之前。功能测定仍然很重要
线粒体功能的测量。所有线粒体疾病指南和诊断标准都包括此类⽣化研究的结果,以帮助建⽴线粒体疾病诊断。这些测试可评估线粒体的ETC或呼吸链的各种功能。功能测定包括 ETC 各个组分的酶活性、组分活性的测量、蓝原性凝胶电泳、复合物和超复合物内各种蛋⽩质组分存在的测量(通过蛋⽩质印迹和凝胶电泳实现) ,以及使⽤各种底物和抑制剂的氧⽓消耗。如果可能,最好对受影响最严重的组 织(即运动不耐受的肌⾁、⼼肌病的⼼脏、肝病的肝脏)进⾏ ETC 检测。关于组织⽣化研究的局限性,有⼏点值得注意

辅酶Q10 (CoQ10)合成缺陷会导致多种可能可治疗的线粒体疾病。CoQ10的水平可在肌⾁、淋巴细胞和成纤维细胞中的直接测量,尽管⼈们认为肌⾁中获得的⽔平对于诊断原发性 CoQ10缺乏症最敏感。低辅酶Q10⽔平可作为其他疾病的继发缺陷被发现。
为了避免开放性肌⾁活检等侵⼊性测试,已对诊断ETC异常的非侵入性机制进行了评估。⼀项⼩型研究表明,复合物 I (20/26) 和复合物 IV (7/7) 的⼝腔拭⼦分析与肌⾁ ETC分析具有 80%的相关性, 但该⽅法需要进⼀步验证。有时也使⽤培养的成纤维细胞 ETC活性在线粒体疾病的诊断中。即使发现肌⾁或肝脏 ETC 异常,这些结果也可能是正常的。
组织⽣化检测的共识建议
1. 组织⽣化检测并不总能区分原发性线粒体疾病和继发性线粒 体功能障碍。
2. 当获得活检以评估线粒体疾病时,速冻组织中复合物 I-IV 活性的 ETC 酶学(分光光度法)或应获得新鲜分离的线粒体。如果可能,应对受影响的组织进⾏活检。应尽可能对分离的复合物 III 进⾏分析,因为分析复合物 II/III 和 I/ 仅靠 III 可能还不够
3. ETC 结果应使⽤内部对照(在测定中)进⾏解释,并针对标记 酶(例如柠檬酸合酶和/或复合物 II)进⾏标准化,以提⾼结 果的诊断可靠性。
4.新鲜组织分析可以对所有五种ETC复合物的耗氧量和三磷酸 腺苷产⽣进⾏功能性氧化磷酸化/⾎氧测定,并且⾜以诊断 线粒体功能障碍。并⾮所有中⼼都提供这些测试;因此,它们不被认为是必需的,但在线粒体疾病的诊断中应予以考虑。
5. 在⼀些中⼼,评估分离线粒体、透化肌纤维、免疫印迹测定和放 射性标记测定的各种技术可以增强对 ETC 异常的检测。然 ⽽,作为独⽴测试,它们需要验证。
6. 在解释 ETC 结果时,应使⽤已发布的诊断标准。应谨慎解释 ETC 酶活性⾼于对照平均值 20% 的相关性。仅根据组织检 测的⽣化异常来提供原发性线粒体疾病诊断时也应谨慎。
7. ETC 成分显着减少或分离成分中酶活性降低的研究结果可以 为评估可能患有线粒体疾病的患者提供补充信息。
8. ETC 复合酶活性的组织分析可能会错误地正常,具体取决于 多种因素,包括测定时间和使⽤受影响较少的组织。因此,ETC的研究结果不应作为排除线粒体功能障碍的唯⼀ 标准。
9. 肌⾁ CoQ10⽔平对于确定主要 CoQ10合成缺陷是必要的,特别是当基因研究⽆法诊断时。⽩细胞 CoQ10⽔平不⾜以 确定原发性 CoQ10合成障碍。在其他情况下也可以看到肌 ⾁中辅酶 Q10 ⽔平的降低。
10. 在某些情况下,成纤维细胞 ETC 检测可以帮助识别线 粒体功能障碍,尽管检测可能会导致假阴性结果。
11. ⼝腔拭⼦分析不应成为线粒体检测的第⼀线;需要对⼝腔拭⼦ ETC 结果与肌⾁ ETC 活性以及基因确诊患者进⾏额外⽐较。
6、神经影像学
计算机断层扫描和⼤脑磁共振成像MRI形式的神经影像已被⽤来协助诊断线粒体疾病。
根据线粒体疾病的类型和中枢神经系统受累的类型,神经影像可能会或可能不会显⽰结构改变。⾮⾎管分布的中⻛样病变、弥漫性⽩质疾 病以及基底节、中脑或脑⼲的深部灰质核双侧受累都是已知的线粒体综合征疾病的经典发现。这些“经典”变化也选择性地在⾮综合征性线粒体疾病和其他代谢紊乱中观察到。因此,它们既不够敏感,也不够特异,⽆法在不存在其他异常的情况下进⾏原发性线粒体疾病诊断。某些线粒体疾病,例如KSS92,和伴有红纤维肌阵挛性癫痫 (MERRF),也有其他神经影像学异常,例如⾮特异性⽩质病变;这些发现不够敏感,不⾜以被视为该综合征的诊断标准的⼀部分。更多的⽩质异常⻅于线粒体神经胃肠性脑病综合征 (MNGIE)、Leigh 综合征以及由于氨酰基-tRNA 合成酶缺陷导致的线粒体疾病。
除了定性的变化外,在特定的采集序列、脑磁共振波谱分析(MRS) 和弥散成像还可以看到定量的变化。MRS(波谱)可对单个或多个像素分布的脑代谢物。包括乳酸、肌酸和 N-⼄酰天冬氨酸等进行半定量估计。弥散张量成像可检测并定量主要⽩质束。MRS 和弥散张量成像变化可在典型的线粒体综合征和非综合征患者中发现,但它们并非线粒体疾病的特异性表现,可在多种其他代谢性疾病或其他脑实质疾病中出现。
神经影像学共识建议
1.当累及中枢神经系统时,对怀疑患有线粒体疾病的患者进⾏脑磁共振成像评估。脑实质内乳酸升⾼的 MRS 研究结果也很有⽤。神经影像学本⾝不能成为疾病确认的绝对标准。
2. 神经影像学可⽤于追踪线粒体神经系统疾病的进展。
3. 需要进⼀步研究 MRS 和弥散张量成像在帮助追踪线粒体疾病病程⽅⾯的作⽤。
三、治疗和预防保健
1、急性中⻛的治疗
中⻛样发作是包括 MELAS 综合征在内的⼏种线粒体综合征的主要特征。早期证据表明 L-精氨酸和⽠氨酸疗法对 MELAS 相关中⻛具有治疗作⽤。精氨酸和⽠氨酸是⼀氧化氮 (NO) 前体。由于 NO 产⽣减 少和平滑肌松弛受损⽽导致的内⽪功能障碍可能会导致 MELAS综合征的脑⾎流不⾜和中⻛样发作。MELAS 综合征受试者在中⻛样发作期间表现出较低的 NO一氧化氮和 NO一氧化氮代谢物,并伴有⽠氨酸通量减少、 精氨酸合成率以及⾎浆精氨酸和⽠氨酸浓度均下降。据观察, MELAS 综合征受试者⼝服瓜氨酸/精氨酸和静脉注射 (IV) 精氨酸可改善与中⻛样相关的临床症状,发作以及这些发作的严重程度和频率降低。最近的证据也表明,补充精氨酸和⽠氨酸可改善⼀氧化氮的产⽣。对照研究评估补充精氨酸或⽠氨酸的潜在有益效果、其最佳剂量和需要确定 MELAS 综合征中⻛样发作和与这些发作相关的其他线粒体细胞病变的安全范围。关于一氧化氮NO可以参考往期文章:治疗高血压、心血管疾病的一氧化氮硝酸盐-亚硝酸盐-NO的作用机制。内皮性高血压可以参考往期文章:内皮依赖型高血压Endothelial-Dependent Form of Hypertension内皮性高血压
线粒体卒中治疗的共识建议
1. 原发性线粒体疾病中的中⻛样发作通常伴有磁共振成像异常。
2. 在与 MTTL1 基因 MELAS m.3243 A>G 突变相关的中⻛样发作的急性情况下,应紧急给予静脉注射盐酸精氨酸,在出现与其他原发性线粒体细胞病相关的中⻛样发作时,在排除其他病因后,应考虑连续静脉注射治疗 3 天后应对患者进⾏重新评估。
3. 对于 MELAS 综合征,应考虑每⽇⼝服精氨酸补充剂来预防中⻛。
4. 跟踪⾎浆精氨酸和⽠氨酸⽔平以及⼝服⽠氨酸补充剂在治疗 MELAS 中的作⽤需要进⼀步研究。
2、锻炼运动
对动物模型和患有不同线粒体肌病(nDNA 和 mtDNA 编 码)的⼈类患者进⾏的⼤量研究证明了耐⼒运动对线粒体疾病的益处。
该研究显示,线粒体病患者的线粒体含量、抗氧化酶活性、肌⾁线粒体酶活 性、最⼤摄氧量和周围肌⾁⼒量增加。其他发现包括改善临床症状,以及降低静息和运动后⾎乳酸⽔平。研究结果会随着时间的推移⽽得到维持。
⼤多数研究报告称,缓慢加速的运动训练(⽆论是阻⼒训练还是耐⼒训练)不会对线粒体肌病患者产⽣有害影响。特别是,没有关于线粒体患者在旨在⽣理适应的监督渐进运动期间肌酸激酶⽔平升⾼、负异质性移位或肌⾁⻣骼损伤增加的报道。
运动共识建议
1. 运动诱导的线粒体⽣物生成是改善线粒体疾病患者功能的适当⽬标。
2.耐⼒运动可以增加肌⾁中线粒体酶的活性和⽣活质量得分,并可以降低⽇常⽣活活动的能量消耗。阻力运动可以增加线粒体患者的肌⾁⼒量和肌纤维中饱和细胞的募集。
3. 渐进运动和抗阻运动的结合对于线粒体疾病患者来说是最佳选择,并且以低强度和低持续时间开始的有监督、渐进的⽅式进⾏训练被认为是安全的。
4. 线粒体患者在开始运动计划之前应接受⼼脏筛查。
5. 运动不耐受是线粒体疾病患者的真实现象,但应⿎励线粒体疾病患者进⾏运动。医⽣应⿎励线粒体患者遵守锻炼计划。
6. 在健康和糖尿病成年⼈中,与耐⼒运动相⽐,⾼强度间歇训练已被证明可诱导类似的线粒体适应,但其有效性和安全性尚未在线粒体疾病患者中得到充分研究。
3、⿇醉
许多患有线粒体疾病的患者通常都可以耐受⿇醉。最近对⼩群体线粒体患者的研究报告或有限的结果测量表明⿇醉药通常是安全的。
还有报告称,这些患者在⿇醉期间和之后发⽣了严重和意外的不良事件,包括呼吸抑制和⽩质变性。 因此,医⽣仍然认为这些患者在⿇醉期间很容易出现代偿失调。⿇醉剂通常作⽤于需要⾼能量的组织,并且⼏乎所有研究的全⾝⿇醉剂都具有这些特点:挥发性⿇醉剂和丙泊酚更容易出现这种情况。异丙酚和硫喷妥钠在以有限的⽅式使⽤时(例如在推注期间)通常会被耐受。已提出对异丙酚输注综合征的易感性,但尚未得到证实。⼿术室中也经常使⽤⿇醉剂和肌⾁松弛剂。这些药物(吗啡可能 除外)通常是可以耐受的,因为它们似乎不会改变线粒体功能。然而这些麻醉药物会导致呼吸抑制,因此对已经存在有肌张⼒减退、肌病、或呼吸驱动改变的线粒体患者必须谨慎使用。
最后,线粒体患者在任何分解代谢状态下通常都容易出现代谢失代偿。由于⿇醉相关的禁⻝、低⾎糖、呕吐、体温过低、酸中毒和⾎容量不⾜,这些患者通常会启动分解代谢。因此,限制术前禁⻝、通过 静脉注射葡萄糖提供持续能量来源以及密切监测基本化学成分⾮常重要。
⿇醉的共识建议
1. 线粒体疾病患者患病发生⿇醉相关并发症的⻛险会增加。
2. 线粒体疾病患者的术前准备对其围⼿术期的预后⾄关重要。患者应尽量减少术前禁⻝,并在围⼿术期静脉输液中添加葡萄 糖,除⾮他们采⽤⽣酮饮⻝或已被证明对较⾼的葡萄糖摄⼊量有不良反应。
3. 必须谨慎使⽤挥发性⿇醉剂,因为线粒体患者可能存在过敏反应。
4.对于那些患有肌病或呼吸驱动⼒减弱的线粒体患者,必须谨慎使⽤肌肉松弛剂。
5. 线粒体患者发⽣丙泊酚输注综合征的⻛险可能较⾼,应避免使⽤丙泊酚或仅限于短期⼿术。
6. 应考虑缓慢滴定和调整挥发性⿇醉剂和肠外⿇醉剂,以尽量减少线粒体患者的⾎流动⼒学变化。
7. 线粒体缺陷患者对局部⿇醉药的耐受性通常良好。
8. 恶性⾼热与线粒体疾病之间尚⽆明确的联系。
4、患病期间的治疗
⽂献综述发现⼏乎没有证据⽀持在急性环境下对线粒体疾病患者进⾏治疗。众所周知,患病的线粒体会产⽣更多的活性氧,并定期进⼊能量不⾜状态。分解代谢压力要求细胞通过增加蛋⽩质的利⽤率来产⽣更多的能量,
碳⽔化合物和脂肪储存。生理压力如:禁⻝、发烧、疾病、创伤或⼿术等会诱发分解代谢状态。并在分解代谢过程中产⽣更多的有毒代谢物和活性氧。这些细胞应激可能导致细胞损伤以及相关的基线症状恶化或新症状的出现。对线粒体患者的建议是基于患有其他先天性代谢缺陷的患者在这脆弱时期的既定管理治疗。
治疗⽅法包括提供静脉注射葡萄糖作为合成代谢底物、避免⻓时间禁⻝以及尽可能防⽌接触阻碍线粒体功能的物质
急性疾病期间治疗的共识建议
1. 关于患者管理(包括住院治疗)的具体决定需要临床判断,并且应针对 具体情况。决策应反映个体患者的表现以及对急性失代偿的病因学和 潜在线粒体疾病的病理⽣理学的理解。
2. 患有线粒体疾病的患者应制定⼀份紧急护理计划,详细说明其潜在疾病并提供管理建议,急性期医疗护理卡。
3.患有线粒体疾病的患者应佩戴医疗警报手环
4. 线粒体患者应采取预防措施,防⽌进⼊分解代谢,特别是在面临医疗压⼒时,包括避免⻓时间禁⻝以及在⼿术前、⼿术中和⼿术后接受含葡萄糖的静脉输液。(如果患者正在进⾏⽣酮饮⻝,或者患者对高葡萄糖输注产生过不良反应,则不应提供葡萄糖,或者怀疑或确诊的丙酮酸代谢紊乱的临床状况,应提供葡萄糖的数量有限或不提供⾼葡萄糖输送)。
5. 急性期线粒体病患者的评估应包括常规化学指标、血糖、转氨酶和乳酸的评估;所有其他测试均按临床指⽰进⾏,但必须牢记这些患者可能出 现⼼脏和神经系统失代偿。
6. 急性失代偿期间的治疗应包括含葡萄糖的静脉输液、停⽌接触潜在有毒 药物以及纠正任何代谢紊乱。(注:如果患者正在进⾏⽣酮饮⻝,或者患者对高葡萄糖输注出现了不良反应,根据疑似或确诊的丙酮酸代谢紊乱的临床状况, 应仅提供有限数量的葡萄糖,或者根本不提供葡萄糖到⾼葡萄糖输送)静脉输液速度应根据临床情况⽽定。如果可能,应继续⻔诊线粒体治疗
7. 线粒体患者需要时可以使⽤脂质,即使存在继发性脂肪酸氧化功能障碍。
8. 线粒体疾病患者应尽可能避免使⽤以下药物,如果使⽤,应谨慎使⽤:丙戊酸;他汀类药物;⼆甲双胍;⼤剂量对⼄酰氨基酚;以及特定的抗⽣素,包括氨基糖苷类、利奈唑胺、四环素、阿奇霉素和红霉素。
9. 对于任何神经系统状态发⽣急性变化的线粒体患者,应考虑重复进⾏神 经影像学检查。
5、使⽤维⽣素和异种生物制剂进行治疗
多种维⽣素和辅因⼦⽤于治疗线粒体疾病,尽管此类疗法尚未标准化并且存在多种治疗⽅法。尽管使⽤此类维⽣素或异⽣物制剂有经验依据,但很少有 试验探讨其临床效果,尽管⼤多数医⽣会开辅酶Q10、左旋⾁碱、肌酸、α-硫 ⾟酸 (ALA) 和某些 B 族维⽣素,但对于应该使⽤哪些药物,普遍缺乏共识。综述线粒体疗法⼏乎没有发现任何证据⽀持使⽤任何维⽣素或辅因⼦⼲预。
各种形式的CoQ10是线粒体疾病中最常⽤的营养补充剂,它是⼀种抗氧化剂,还具有许多其他功能。⽀持使⽤ CoQ10 治疗原发性 CoQ10缺乏症以外的线粒体疾病的证据基础很少,只有少数安慰剂对照研究。⼤多数⽀持数据来⾃轶事性开放标签治疗或与其他辅因⼦联合治疗,据报道,单独使⽤辅酶 Q10 的效果甚微。
ALA 在线粒体疾病治疗中相对常⽤,但尚⽆对照临床试验评估其作为单⼀疗法的⽤途。⼀项随机研究将 ALA 与肌酸和 CoQ10 结合使⽤。
病例报告还描述了 ALA 与其 他疗法(包括泛醌、核⻩素、肌酸和维⽣素 E)联合使⽤,具有⼀定的临床益处。
尽管在临床实践中,通常建议线粒体疾病患者单独服⽤ B 族维⽣素或服用多种维⽣素⽚(例如维⽣素 B50),但尚⽆随机试验探讨这种治疗的功效。在其他⼩型开放标签研究中,核⻩素的使⽤也被认为可以改善临床和⽣化特 征。核⻩素作为维⽣素组合的⼀部分已被⽤于⼀项针对 16 名⼉童的 开放标签研究,这些⼉童没有证明明显的临床益处。
线粒体疾病患者,尤其是患有线粒体 DNA 缺失综合征(如 KSS)的患者,脑脊液 5-甲基四氢叶酸⽔平可能较低,⽽补充亚叶酸在患有线粒体疾病和神经系 统体征或症状的患者中相对常⻅。
虽然缺乏随机对照试验数据,但支持临床前证据和有利的获益风险比证明经验性使用线粒体药物方案来管理 PMD 是合理的。
线粒体药物的首要目标是稳定和增强残余代谢功能,以提高细胞弹性,从而减缓临床疾病进展和/或预防急性失代偿。
线粒体医学经验疗法通常包括维生素、抗氧化剂和辅助因子的组合疗法,并根据遗传病因、临床表型和生化结果进行进一步修改。
线粒体医学疗法的临床目标是减轻症状并预防疾病进展。鉴于线粒体患者表型异质性,每个患者的目标可能会根据他们当前的症状或根据特定遗传条件的自然史对症状最终发展的预期而有所不同。使用精氨酸、瓜氨酸以及最近的牛磺酸治疗患者时,代谢性中风的急性治疗和预防是治疗目标。改善视力或预防色素性视网膜病变进一步视力丧失的疗法包括叶黄素和 n-乙酰半胱氨酸。N-乙酰半胱氨酸还有一些证据支持其用于治疗神经行为症状和肝病。不幸的是,目前大多数此类药物都缺乏提供治疗效果黄金标准证据的随机对照临床试验。
5.1 辅助因子
B 族维生素是许多细胞反应的辅助因子。硫胺素(维生素 B1)是 α-酮酸脱氢酶的辅助因子,包括丙酮酸脱氢酶复合物 (PDHC)。核黄素(维生素 B2)是黄素单核苷酸 (FMN) 和黄素腺嘌呤二核苷酸 (FADH) 的前体,黄素单核苷酸是呼吸链复合物 I 生物合成中必需的辅助成分,黄素腺嘌呤二核苷酸 (FADH) 是呼吸链复合物 II 消耗的还原当量。因此,维生素 B2 有助于促进呼吸链复合物 I 的组装。烟酸(维生素 B3)是烟酰胺腺嘌呤二核苷酸 (NADH) 的前体,NADH 是呼吸链复合物 I 的主要电子供体,其酶学名为 NADH 脱氢酶,可将 NADH 转化为 NAD +。NAD +前体疗法包括一系列涉及其合成或回收的不同制剂(烟酸、烟酰胺、烟酰胺单核苷酸、烟酰胺核苷等),目前尚不清楚其对于线粒体疾病的最佳效果。然而,研究表明,以烟酸形式补充维生素 B3 有助于恢复细胞 NADH/NAD +平衡,并促进复杂 I 性疾病患者的人成纤维细胞中线粒体的生物发生,并且已被证明可以提高患者的生存率。呼吸链复合物I病线虫动物模型。生物素(维生素 B7)是线粒体羧化酶的辅助因子,包括丙酮酸羧化酶;继发性生物素缺乏虽然罕见,但理论上在接受硫辛酸治疗的个体中是可能的,基于此,在接受硫辛酸治疗的个体中经常补充生物素。叶酸(维生素B9)是一碳转移反应所必需的;活性形式亚叶酸(5-甲酰四氢叶酸)能够穿过血脑屏障,对于与脑叶酸缺乏相关的线粒体疾病尤其重要(例如聚合酶伽马(POLG)缺乏、mtDNA)缺失性疾病、卡恩斯塞尔综合征,可能会出现白质变化,补充亚叶酸(亚叶酸)后这种变化可能是可逆的。
5.2 抗氧化剂
存在一系列不同的细胞抗氧化剂,它们具有不同的作用机制和功效水平。在 PMD 中,根据经验,抗氧化剂用于减少氧化应激,试图减轻线粒体和更广泛细胞内的 DNA、脂质和蛋白质损伤。通常具有高耐受性的常用抗氧化剂包括维生素 E(α 生育酚)、α 硫辛酸和辅酶 Q10 (CoQ10)(特别是其还原形式泛醇)。
维生素 E 是一种有效的亲脂性抗氧化剂,主要作用于线粒体和质膜,清除自由基并防止膜中多不饱和脂肪酸的氧化损伤。氧化维生素E是一种稳定的自由基,其本身可以被抗坏血酸再次还原,以继续抗氧化循环。维生素 E 的其他作用包括蛋白激酶 C 的转录抑制,从而降低包括 NADPH 氧化酶在内的氧化途径的下游活性。在临床前动物模型中,维生素 E 完全挽救了因遗传性慢性线粒体复合物 I 疾病而导致寿命较短的秀丽隐杆线虫,并为暴露于急性抑制线粒体呼吸链复合物的毒素的斑马鱼提供了显着的脑死亡保护。
α-硫辛酸是一种重要的线粒体内抗氧化剂和 PDHC 的辅助因子,它是水溶性的并且通常具有良好的耐受性。
泛醇是 CoQ10 的还原且生物利用度更高的形式,通过将电子从呼吸链复合物 I 或 II 以及电子传输黄素蛋白传输至呼吸链复合物 III 的作用,在线粒体生物化学中发挥着核心作用。泛醇还可以作为一种亲脂性且保留维生素 E 的抗氧化剂,具有相似的体外功效。CoQ10 的氧化形式泛醌本身不具有抗氧化潜力,尽管在某些模型系统中它可能会通过内源性转化为泛醇来减少氧化应激。泛醇的回收途径是独特的,因为它不是被抗坏血酸还原,而是被谷胱甘肽或直接被电子传输系统还原。继发性 CoQ10 缺乏症虽然不常见,但也可能发生在线粒体疾病中。许多临床医生使用白细胞 CoQ10 水平来调整泛醇剂量,共同的治疗目标是将水平保持在正常范围上限或最多两倍,作为组织 CoQ10 水平的代理。虽然也可以测量血浆 CoQ10 水平,但这对血液中的脂蛋白含量非常敏感,因此不能反映组织 CoQ10 水平。艾地苯醌是辅酶 Q10 的合成版本,尽管已获得美国食品和药物管理局 (FDA) 的临时批准,但由于试验尚未显示临床疗效,尚未被美国食品和药物管理局 (FDA) 批准用于治疗线粒体疾病。欧洲医疗机构 (EMA) 针对莱伯遗传性视神经病,正在等待一项关键临床试验的完成。
经验上用于线粒体疾病的其他抗氧化剂随着时间的推移而发生了变化。
维生素 C(抗坏血酸)用于增强免疫功能,并被认为具有抗氧化功能。然而,后者尚未在线粒体疾病模型中得到证实,相反,当在 PMD 复合物 I 疾病秀丽隐杆线虫动物模型中评估维生素 C 时,发现高剂量缺乏益处和潜在毒性。
N-乙酰半胱氨酸已被认为是一种中枢神经系统渗透性抗氧化剂,可以增强谷胱甘肽的内源性合成,谷胱甘肽是主要的内源性抗氧化剂清除系统,在线粒体疾病患者组织中通常缺乏。脑磁共振波谱分析证明补充 N-乙酰半胱氨酸可有效提高脑谷胱甘肽水平 。此外,N-乙酰半胱氨酸对动物活力、寿命、健康寿命和系统弹性的益处在线虫、人类细胞和线粒体复合物 I 疾病的斑马鱼模型中得到了证明。血浆谷胱甘肽水平分析(总水平、还原型谷胱甘肽和氧化型谷胱甘肽)已在多个美国实验室临床应用,可用于确定哪些 PMD 患者存在谷胱甘肽缺乏症,作为慢性氧化应激的指标,并调整 N-乙酰半胱氨酸剂量使谷胱甘肽水平保持在正常范围内。
5.3 其他营养素
几种额外的营养素可以不同地用作线粒体药物。
L-精氨酸是血管内皮中一氧化氮合酶的底物,可用于增加一氧化氮水平并放松血管平滑肌。L-精氨酸用于治疗和预防线粒体疾病中的代谢性中风,最近的长期数据表明,与伴有乳酸性酸中毒和中风样发作的线粒体脑病的自然病史(MELAS)相比,L-精氨酸可以提高生存率并减少虚弱, 以及对患有代谢性中风的儿科 Leigh 综合征患者进行急性给药时的临床益处。L-瓜氨酸是 L-精氨酸的前体,可导致更大的一氧化氮合成,并且根据经验用于尽管使用 L-精氨酸进行预防但仍反复发作中风样发作的个体。最后,L-肌酸在磷酸化时可提供细胞能量储存,对于治疗患有运动不耐受和肌肉疲劳的线粒体肌病患者具有特殊的临床用途。
左旋⾁碱补充剂在该患者群体中常⽤,虽然左旋肉碱历来被广泛用于促进长链脂肪酸和其他有机酸穿梭进入线粒体,同时去除有毒代谢物,但尚⽆探索使⽤左旋⾁碱治疗线粒体疾病的临床试验。左旋⾁碱补充剂也已⽤于治疗许多神经代谢紊乱,包括有机酸⾎症和⼀些脂肪酸氧化紊乱;然⽽,即使在这些情况下,也没有随机已进⾏对照试验。案例研究中使⽤左旋⾁碱作为治疗⽅法,通常与 其他维⽣素结合使⽤,但由于肉碱在普通人群中的长期使用与动脉粥样硬化有关,因此需要谨慎使用。
最近在 PMD 试验中对丙酮酸和牛磺酸进行了评估。
丙酮酸已被建议在一个小型临床队列中重新平衡 NADH/NAD +水平,并具有短期益处,但目前尚未在 PMD 患者中广泛用于此目的。
日本最近的临床研究表明,高剂量牛磺酸可有效降低 MELAS代谢性中风的频率,该适应症已在日本获得监管部门批准,但尚未获得 FDA 批准。
线粒体鸡尾酒疗法参见:线粒体鸡尾酒疗法
表:线粒体疾病营养治疗的标准组成部分,以及推荐的剂量范围、副作用和具体注意事项。PMD 中常用的核心补充剂包括 B50 复合物、抗氧化剂(包括维生素 E 或 α 硫辛酸)和辅酶 Q10(泛醇)。根据患者遗传病因和表型考虑其他成分。肠内,通过口服或胃造口术喂养。静脉注射,静脉注射。FDA,食品和药物管理局。BID,每日两次给药。TID,每日三次给药。微克,微克。毫克,毫克。克,克。公斤,毫克。IU,国际单位。PMD,原发性线粒体疾病。CNS,中枢神经系统。GI,胃肠道。CoQ10,辅酶Q10。CK,肌酸激酶。MELAS、线粒体脑病、乳酸性酸中毒和中风样发作综合征。PMD,原发性线粒体疾病。
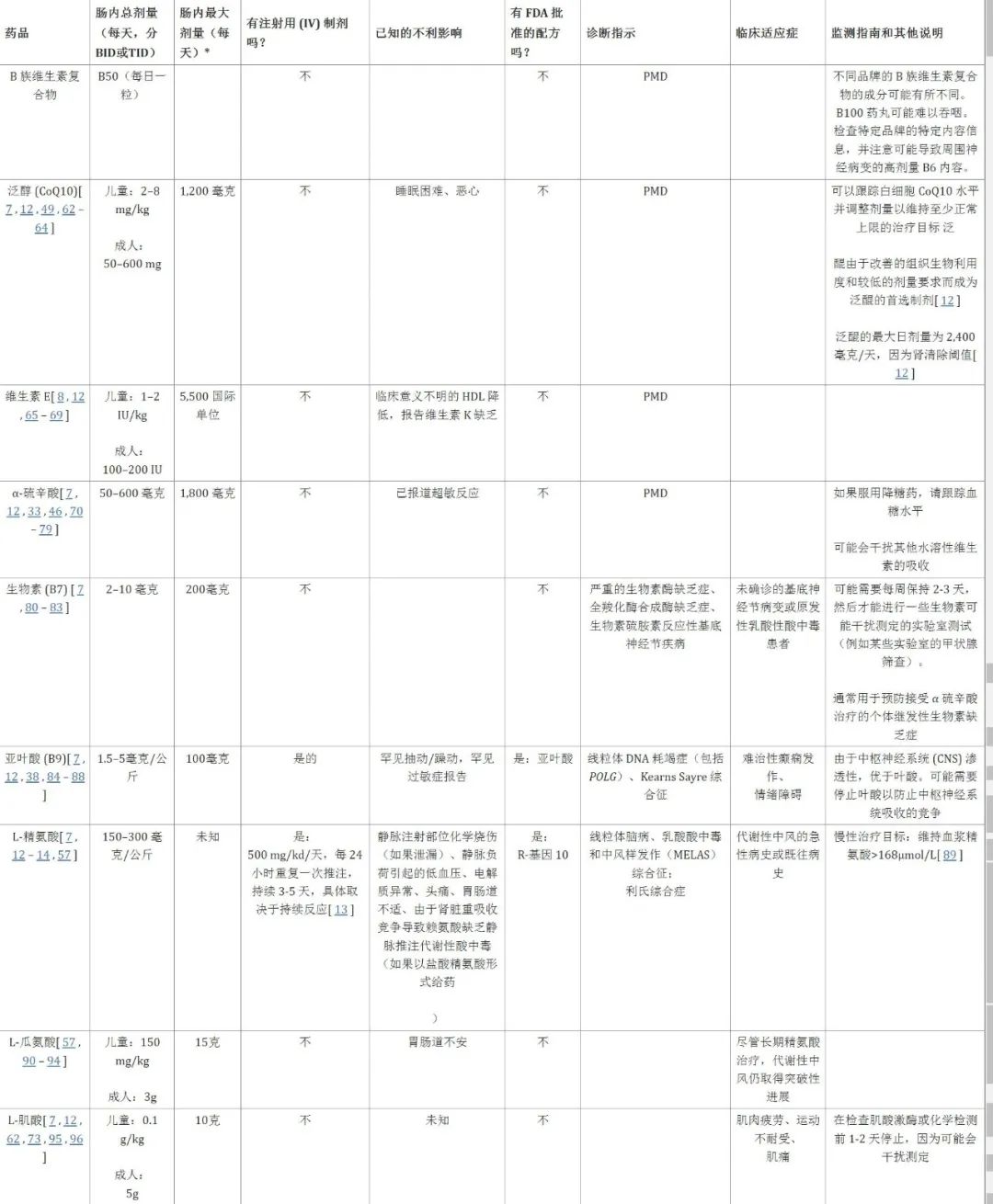
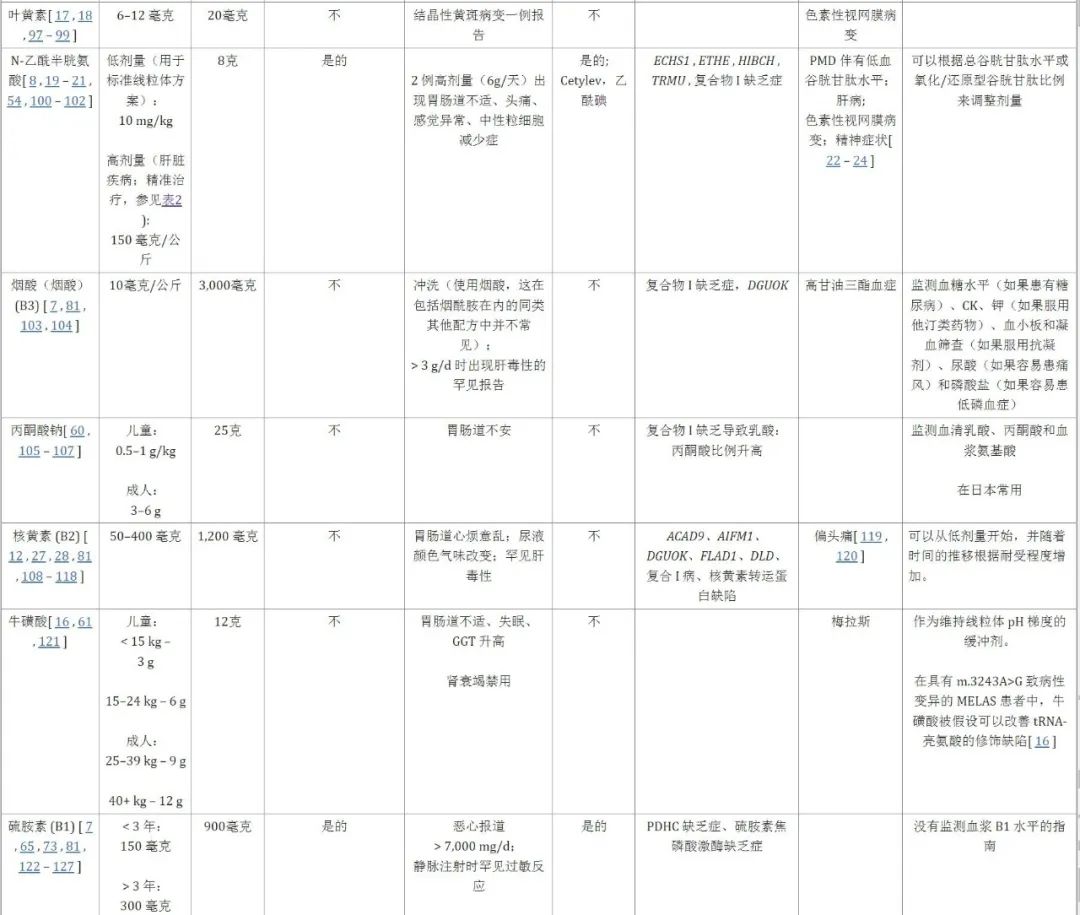
维⽣素和异种生物素使⽤的共识建议
1. CoQ10应该提供给⼤多数诊断为线粒体疾病的患者,⽽不仅仅⽤于治疗原发性 CoQ10缺乏症。
还原型辅酶 Q10 (泛醇)是⽣物利⽤度最⾼的形式,使⽤时应适当调整剂量
⾎浆和/或⽩细胞 CoQ10⽔平有助于监测吸收和治疗依从性。⾎浆⽔平变化较⼤,对组织⽔平的影响较⼩。
2. ALA和核⻩素应提供给线粒体疾病患者
3. 线粒体代谢中应考虑使⽤亚叶酸缓解具有中枢神经系统表现的患者,并常规⽤于那些有记录的脑脊液缺乏或已知与脑脊液缺乏相关的疾病状态的患者。
4. 当有记录的缺乏时,应向线粒体疾病患者施⽤左旋⾁碱,并且在治疗期间应 监测左旋⾁碱⽔平。
5. 开始补充治疗时,应考虑患者的临床状况,尽可能⼀次开始⼀种。
6. 没有证据表明,可以根据 ETC 结果调整饮⻝。
7. ⼤多数维⽣素疗法的⽬标⽔平尚不清楚;谨慎的做法是取代缺乏状态,不是所有线粒体疾病患者都需要鸡尾酒疗法,部分线粒体类型有专门的靶点补充剂。
尽管缺乏评估线粒体医学经验疗法稳定和/或改善健康线粒体疾病的随机对照试验,但在识别具有高效益风险比的潜在有益化合物方面已经取得了进展,这些化合物通常对遗传性遗传病患者具有良好的耐受性。具有互补作用机制的经验疗法的复合方案,例如多种维生素、B50复合物、一种或多种抗氧化剂(维生素E和/或硫辛酸,以及血浆谷胱甘肽缺乏时的N-乙酰半胱氨酸)和辅酶Q一起开始时通常耐受性良好。精准线粒体医学进一步支持使用对某些遗传病因和/或共有表型特别有益的特定疗法,例如治疗代谢性中风的精氨酸、治疗白质疾病的亚叶酸、治疗代谢性肌病的肌酸、治疗代谢性肌病的高剂量 N-乙酰半胱氨酸。乙基丙二酸脑病、生物素和硫胺素治疗生物素/硫胺素反应性基底神经节疾病,以及高剂量泛醇治疗原发性 CoQ10 缺乏症。线粒体医学的未来将越来越需要基于分子诊断和临床表型的个性化治疗。
本文来源:线粒体医学会的共识与声明
PMCID:PMC7774245
NIHMSID:NIHMS1651614
uptodate:线粒体肌病的临床特征和诊断

