1 背景知识介绍
在进行PCR反应时,寡核苷酸引物(Oligonucleotide primers)是必要的。我们需要设计与DNA/ cDNA模板区互补的引物。一次添加一个核苷酸时,必须选择性地阻断(selectively block)和解封(unblock)核苷酸上的反应性基团(reactive groups)。引物的主要特性是它们必须与模板分子上的序列相对应(必须与模板链互补(complementary to template strand))。
当然,引物不需要与模板链完全对应(例如添加酶切位点或者是构建突变位点的引物);但引物的 3'端必须与模板 DNA 链完全对应(因此我们添加酶切位点均在5'端),这样才能进行延伸。通常在 3' 端使用鸟嘌呤(guanine, G)或胞嘧啶(cytosine, C),尽量不要出现腺嘌呤(adenine, A),引物的 5' 端通常有几个核苷酸的延伸(stretches)。此外,杂交引物(hybridized primers)的 3' 端必须相互指向,因此我们在合成引物的时候均是从5'端到3'端。
引物的长度对于扩增特异性序列很重要。短引物主要用于扩增小而简单的DNA片段。另一方面,长引物用于扩增真核生物基因组DNA样本。然而,引物不应该太长(> 30bp引物)或太短(<15bp),当然我们的模板很纯的时候,引物可以不受这些规矩的限制,可以根据实际情况去定义引物的长度。短引物不准确性更高,换句话说就是非特异性的DNA扩增产物增加,表现在电泳时候出现很多非特异性条带,而长引物的弊端在于导致较慢的杂交速率。平均而言,需要扩增的DNA片段的大小应在1- 10kb以内,实践告诉我们,我们比较号扩增的长度在3Kb及以下,当然也跟物种有关。
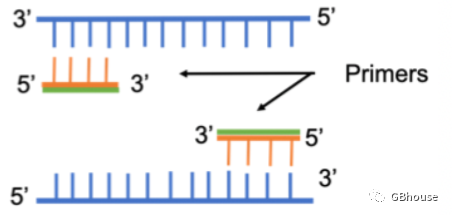
(图1 引物扩增示意图。 PCR基因扩增方向和体内合成方向是一致的,即从5'端到3'端。)
引物设计也应该注重以下规则:引物的结构应相对简单,不含内部二级结构,避免内部折叠;我们还需要避免引物-引物退火,它会产生引物二聚体并破坏扩增过程;在设计时,如果不确定在引物的某个位置放置什么核苷酸,可以在该位置包含多个核苷酸,称为混合位点(mixed site);还可以使用基于核苷酸的分子插入物(肌苷,inosine)来代替常规的核苷酸,以获得更广泛的配对能力。
考虑到上述信息,引物一般应具有以下属性:
(1)长度为18-24个碱基;
(2)G/C含量40-60%;
(3)以1-2对G/C开始和结束;
(4)退火温度(Tm) 50-60℃;
(5)引物对之间的Tm相差应在5°C以内;
(6)引物对不应有互补区域;
(当然这是在比较理想的状态下,如果非理想状态尽量以此作为参考)
2 NCBI引物设计实操
2.1 定量引物规则
最最最重要的是特异性(即只能扩增需要检测的基因),其他规则(1)引物长度为20- 21 bp;(2) 避免引物中连续出现5个或以上相同碱基,例如AAAAA或GGGGG;(3)每条引物两端的碱基最好是G或C(GC碱基之间为三个氢键连接,保证引物与模板连接的稳固性);(4) GC含量为45%~55%;(5)PCR产物大小为85~300 bp;(6)引物尽可能的跨内含子-外显子;(7)参考上节引物设计规则;(8)其它。
2.2 NCBI设计定量引物
2.2.1 NCBI查看mRNA序列(以人TNF-α)为例
(1)检索人类TNF-α基因,方法如图2-1,点击search

(图2-1 NCBI检索人类TNF-α基因)
(2)NCBI定位到需要进行引物设计的基因,左键点击即可
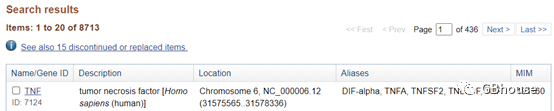
(图2-2 选择我们需要的研究的TNF-α)
(3)TNF-α的相关信息(https://www.ncbi.nlm.nih.gov/gene/7124)
因为我们只为设计引物,过多信息不用看,但是,值得注意的是有的基因拥有很多剪切体,因此选择合适的剪接体是非常重要的,如果知道我们具体研究的是哪个剪切体,直接选择即可;如果不知道我们要研究哪一个剪切体,就选择最长的或者研究最多的剪切体。
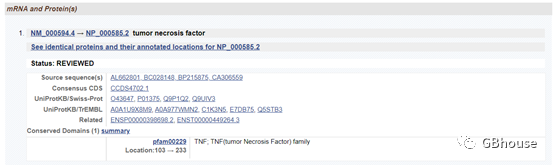
(图2-3 TNF-α剪切体信息,总共1条收录信息,对其进行qPCR引物设计)
(4)点击:NM_000594.4,转到该mRNA的详细信息页面(https://www.ncbi.nlm.nih.gov/nuccore/NM_000594.4)
a. 查看信息包含各外显子、内含子、CDS区域

(图2-4 查看外显子、内含子、CDS区域信息,因为设计的时候尽量跨外显子-内含子)
b. 第一个外显子在1-363,CDS区域在178-897,再借鉴一个设计定量引物规则,一般定量引物会选择mRNA或第一外显子或CDS的前500bp,因此我们这里选择TNF-α前600bp(当然具体多少长度可以适当更改)进行定量引物设计(序列复制或保存为文件均可)。
(5)NCBI引物设计
在这里,我们使用NCBI的BLAST功能选项进行设计
(a)打开NCBI网址(https://www.ncbi.nlm.nih.gov/),找到BLAST选框,下图红框;
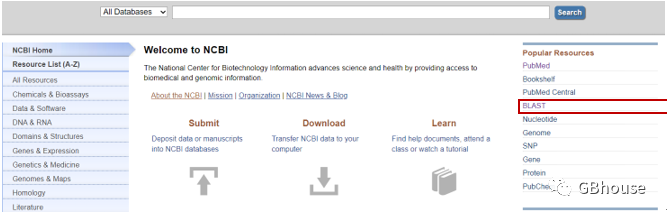
(b)找到“primer- BLAST”选框,并左键单击
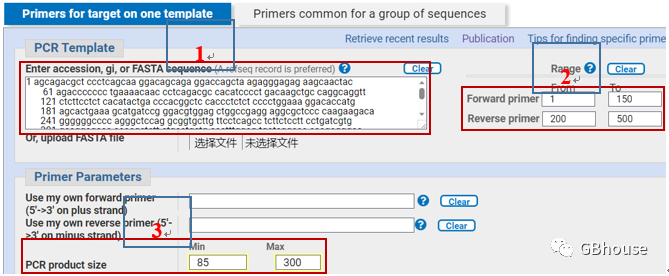
(c)新页面首先:输入碱基序列,或者通过文件拖拽序列进入;接着限定上下游引物的起始位置(记住qPCR产物在85-300bp之间的约定,其它的规则软件会自动设定,不用管),在这里我们选择1-150的位置设定为上游(forward primer)-200-500下游(reverse primer);产物长度设置为85-300,如下图所示。
(d)点击本页面最下边的“get primers”;

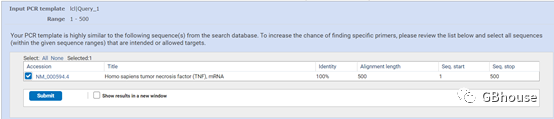
(e)核对并提交的序列隶属的基因信息,复选框打钩,点击“submit”即可;
(f)等待界面,软件设计引物需要时间,数分钟甚至更长,等待……即可;
(g)得到引物信息,这里需要查看引物的特异性,及查看引物下边是否还包含其它的mRNA信息,如果不包含表示引物特异性极高,只能扩增到我们需要的mRNA(TNF-α).
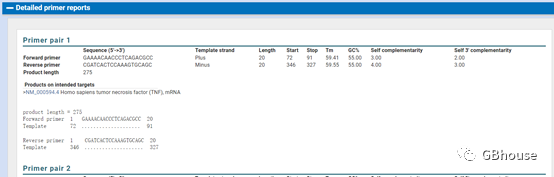
(h)保存所需要的qRT-PCR引物(出现很多条时候,以排名靠前的为主,为了实验尽可能成功,建议合成两对及以上定量引物)
Forward primer: GAAAACAACCCTCAGACGCC
Reverse primer: CGATCACTCCAAAGTGCAGC
3 oligo 7定量引物设计实操
俗话说技多不压身,这个理念对于设计引物也是一个样,极少数情况下NCBI设计的引物在实验中可能不可用,例如溶解曲线不是标准的峰(非单峰)、无法靶向基因,靶向效率低等等。因此,为了实验的成功,在掌握一门其它设计定量引物的办法是极其必要的。对了,文末附上oligo 7的下载位置,不然你们说我耍流氓!
3.1 oligo设计引物规则
具体规则如上所述,回头看看即可,如果还不会把手或者屁股支出来,我打三下,再告诉你这么弄。
3.2 oligo 7设计定量引物
(a)打开oligo 7软件
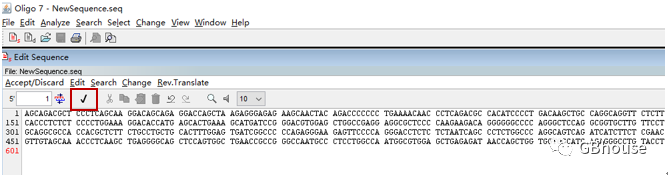
(b)点击:file-new sequence,然后将序列粘贴进去,并点击红框的 “√”即可;
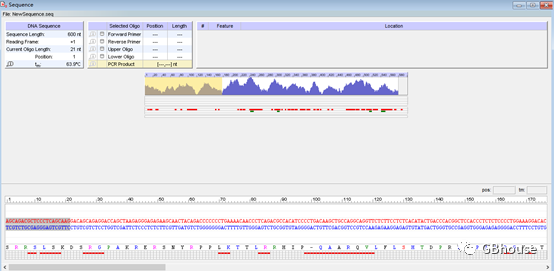
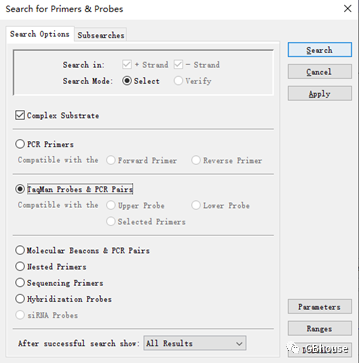
(c)点击“search- for primers & probs…”,然后选择“TaqMan probs & PCR pairs”/当然选择 “PCR primers”也行,不过出现的数量比较多,最后点击“search”即可;
(d)得到引物信息
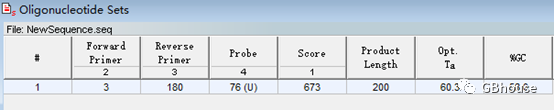
e)保存定量引物,务必选择要保存的引物,“file- save- Forward primer”,自己选好位置(找不到不怪我没说清楚)并命名;
上游引物:

下游引物:

(f)合成引物(导出的引物均是5'端到3'端,直接送合成即可):
Forward primer: CAGACGCTCCCTCAGCAAGGAC
Reverse primer: CCCGGATCATGCTTTCAGTGCTC
(对了打开导出的引物序列需要DNAstar软件,最后在这里也一并送出(windows),下载地址:https://pan.baidu.com/s/1Xd1SGlGo6XxoRTi6NbApLQ;提取码:1179)

