缺血再灌注(IR)损伤是一个涉及众多病理生理过程的复杂级联事件。这些过程包括中性粒细胞、血小板、细胞因子、活性氮物质、活性氧物质(ROS)的激活,凝血系统、内皮以及黄嘌呤氧化还原酶系统的参与。这最终可能导致细胞损伤、细胞死亡、血管通透性增加、组织坏死以及多器官功能障碍。
1.由坏死和凋亡导致的细胞死亡是由缺血再灌注损伤期间释放的物质所引发的。
2.活性氧物质被认为除了在包括肿瘤、动脉粥样硬化和神经退行性疾病等其他几种疾病中发挥核心作用外,在缺血再灌注损伤中也起着核心作用。
3.在人类中,涉及缺血再灌注损伤的最常见综合征包括心肌梗死、心脏搭桥手术以及器官移植。在兽医学中,最常见的综合征包括胃扩张扭转(GDV)、动脉血栓栓塞(ATE)、失血性休克复苏、器官移植、膈疝、头部外伤、肠系膜扭转、肠嵌顿以及脊髓外伤。
我们从以下几个机制来说明IR的可能的机制,要注意IR的机制非常非常广泛,目前医学完全没有办法诠释的清楚,我们只做常见的进行解释。
什么是自由基?
自由基是一类外层电子轨道上存在单个不配对电子的化学物种,涵盖原子、原子团和分子。其种类主要包括以下几种:
氧自由基(OFR):其中具有代表性的是超氧阴离子,它是氧分子在特定条件下获得一个电子后形成的,在许多生理和病理过程中发挥重要作用,例如在细胞呼吸过程中会少量产生,在炎症反应、缺血再灌注损伤等病理情况下会大量生成。
脂性自由基:如烷自由基和烷氧自由基等。这些自由基主要在脂质过氧化过程中产生,当细胞内的脂质受到氧化应激时,不饱和脂肪酸的碳氢键容易发生断裂,从而产生脂性自由基,它们能够引发脂质过氧化的链式反应,进一步破坏细胞膜等生物结构的完整性,影响细胞功能。
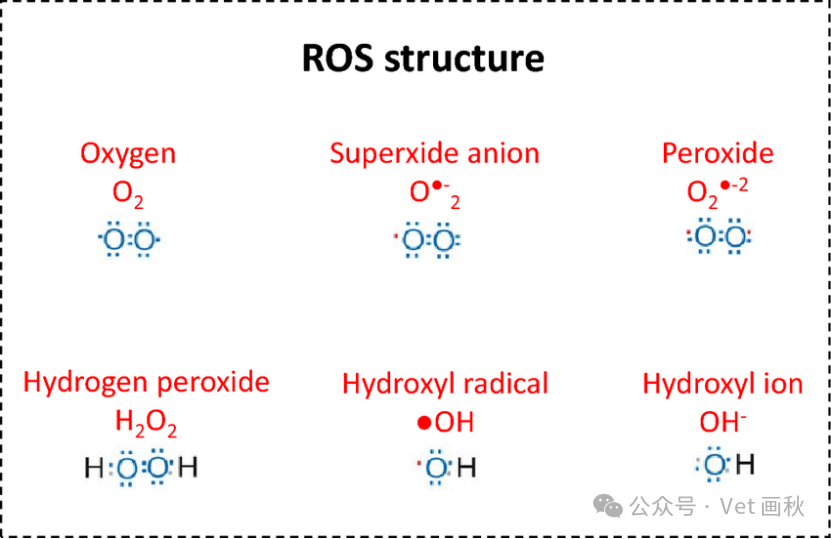
自由基具有高度的化学反应活性,这是由于其未配对电子的存在使其倾向于从周围环境中获取或失去电子以达到稳定状态。在生物体内,这种高反应活性使得自由基既可以参与正常的生理过程,如信号转导等,但如果其产生和清除失衡,过量的自由基会攻击生物大分子,如蛋白质、核酸和脂质,导致细胞损伤,进而引发多种疾病,如心血管疾病、神经退行性疾病、癌症等。因此,维持体内自由基的平衡对于生物体的健康至关重要。
机体本身存在抗氧化防御系统来清除自由基,包括酶类抗氧化剂(如超氧化物歧化酶、过氧化氢酶、谷胱甘肽过氧化物酶等)和非酶类抗氧化剂(如维生素 C、维生素 E、谷胱甘肽等),它们协同作用,使自由基的产生和清除处于动态平衡。
所以自由基就像是一群调皮捣蛋的小家伙,它们的外层电子轨道上有个单个不配对的电子,这让它们特别不安分,总想从周围找点电子来配对,好让自己稳定下来。在我们的身体里,它们有时候也会干点好事,比如参与身体里一些正常的信号传递工作以及白细胞通过释放自由基来破坏入侵的致病微生物,帮助杀死寄生虫、细菌、病毒等。
但是,如果身体里自由基太多了,麻烦就来了。因为它们太活跃了,就会到处去攻击身体里重要的东西,像蛋白质、核酸还有脂质这些大分子。它们这么一捣乱,细胞就会受伤,就可能引发各种严重的疾病。
不过我们的身体也很聪明,有一套自己的 “防御部队” 来对付这些自由基。这里面有一些酶类抗氧化剂,像超氧化物歧化酶、过氧化氢酶、谷胱甘肽过氧化物酶等等,它们可以把自由基变得无害。还有一些非酶类抗氧化剂,比如我们常听说的维生素 C、维生素 E、谷胱甘肽,它们也会来帮忙。这些抗氧化剂一起努力,让自由基的产生和被清除的速度保持平衡,这样我们的身体才能健健康康的。要是这个平衡被打破了,自由基太多了,身体就容易出问题啦。所以我们要尽量保持身体里这种平衡,比如通过健康的饮食多摄入一些抗氧化的营养物质。
氧化应激可由酶促来源和非酶促来源产生。常见的酶促来源包括黄嘌呤氧化酶系统、NADPH氧化酶系统、线粒体电子传递链和非偶联型一氧化氮合酶(NOS)系统。非酶促来源是氧化应激的次要来源,包括血红蛋白和肌红蛋白,特别是在肢体损伤时。黄嘌呤氧化酶系统、NADPH氧化酶系统和线粒体电子传递链广泛涉及多个器官的氧化应激,包括肠道、肺、心脏、大脑、肌肉、肝脏、胰腺、胃和肾脏。NOS是肝脏、心脏和主动脉内皮细胞中的主要氧化应激因素。以下各节将详细讨论这些酶系统。
但是,什么是氧化应激?
氧化应激通常是指在生物体内氧化剂的产生超过了体内抗氧化系统的清除能力。氧化剂主要是指活性氧种(ROS)如超氧阴离子、过氧化氢、羟基自由基等。这些氧化剂能够攻击细胞的脂质、蛋白质和核酸,引发一系列的细胞损伤和病理反应。
那么我们下面将要讲解酶促来源的一种:
黄嘌呤氧化酶系统:
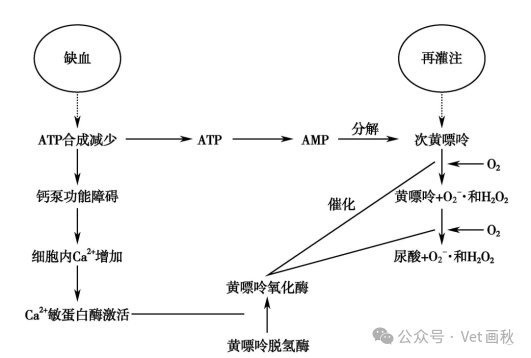
黄嘌呤氧化还原酶在嘌呤代谢中的作用
黄嘌呤氧化还原酶作为一种复杂的钼黄素酶,在嘌呤分解代谢的 “流水线” 上扮演着极为重要的角色,类似于一个精密的 “加工厂”。该酶由黄嘌呤脱氢酶和黄嘌呤氧化酶这两个 “工作模块” 构成,它们协同作业,将次黄嘌呤逐步转化为黄嘌呤,最终生成尿酸。
正常细胞内,嘌呤代谢主要通过两条 “主要通道” 进行。起始阶段,作为众多需能酶促反应中嘌呤供体的三磷酸腺苷(ATP),在脱氨酶的作用下,如同经历了一道 “加工工序”,转化为肌苷一磷酸,随后通过去磷酸化形成肌苷;或者先去除一个磷酸基团转变为腺苷,再进一步转化为肌苷。肌苷如同一个 “中转站”,继续转化为次黄嘌呤,而后被氧化为黄嘌呤。此外,黄嘌呤还可由鸟嘌呤经一系列反应合成而来,鸟嘌呤先转化为鸟嘌呤一磷酸,进而生成黄嘌呤。
在这一复杂的代谢过程中,黄嘌呤氧化酶在工作时会产生一些 “副产品”—— 活性氧(ROS),这些 ROS 如同 “不安分的小粒子”,具有较高的化学反应活性。正常情况下,黄嘌呤脱氢酶在将次黄嘌呤转化为黄嘌呤的过程中,会使辅酶 NAD⁺或分子氧(O₂)发生相应变化,同时产生还原型辅酶 Ⅰ(NADH),整个过程就像一个 “精密的化学反应链”,有条不紊地进行着。
缺血状态下黄嘌呤氧化还原酶的变化及影响
当身体某个部位遭遇缺血状况时,如同 “生产线” 的能源供应出现短缺,细胞内 ATP 水平降低,这一变化就像一个 “信号开关”,触发黄嘌呤脱氢酶发生转变,成为黄嘌呤氧化还原酶。与此同时,关键半胱氨酸的氧化以及有限的蛋白水解作用等因素,如同 “额外的干扰因素”,也会促使黄嘌呤脱氢酶向黄嘌呤氧化还原酶的转变。
当缺血组织的血流恢复,犹如 “生产线” 重新获得动力,黄嘌呤氧化酶开始与 O₂发生反应,以氧作为最终电子受体,促使次黄嘌呤转化为黄嘌呤和尿酸。然而,在此过程中,如同打开了一个 “潘多拉魔盒”,会释放出超氧阴离子(O₂・⁻)和过氧化氢(H₂O₂)等 ROS。这些 ROS 就像一群 “捣蛋分子”,引发氧化应激反应,导致细胞内的氧化还原平衡被打破。它们还会诱导细胞表达如 P - 选择素和细胞间黏附分子 - 1(ICAM - 1)等黏附蛋白,进而引发一系列如同 “多米诺骨牌效应” 般的不良身体反应,最终可能导致组织损伤和疾病的发生。
NADPH氧化酶系统:
坏死与凋亡的基本概念及坏死的传统特征
坏死和凋亡均被视作程序性细胞死亡执行阶段的表现形式,然而,二者对整个生物体产生的影响存在显著差异。坏死作为一种细胞死亡方式,其典型特征表现为早期质膜通透性增加以及细胞器肿胀。当细胞遭受过度的外部压力时,便会触发坏死进程。传统观点认为,坏死是一种被动发生且不受机体调控的细胞死亡过程。尽管在病理学领域,坏死现象广泛存在,但针对缺血-再灌注损伤中坏死调控机制的研究却相对匮乏。坏死通常由物理、化学或生物性损伤所引发的外部环境急剧变化所致,细胞解体、器官肿胀以及线粒体功能丧失构成了坏死的主要特征,并且这一过程会在缺血组织内引发大量局部炎症反应。尽管坏死在多种病理生理过程中均有被观察到,但长期以来,传统观念一直认为坏死不受机体精细调控。
坏死性凋亡:一种新型的程序性细胞死亡形式
然而,近年来在炎症和缺血-再灌注损伤的小鼠模型研究中,证实了坏死存在一种特殊特征,即坏死性凋亡。坏死性凋亡属于程序性细胞死亡范畴,其进程由死亡信号所调控,并呈现出与传统坏死相似的细胞死亡模式。局部缺血状况的产生源于组织血流受阻,这会导致氧气及营养物质供应受限,若这种状态持续时间过长,将会损害细胞能量代谢,进而引发细胞死亡。而在失血恢复(再灌注)阶段,氧气重新引入组织的同时会伴随活性氧(ROS)的产生,从而导致与细胞失活相关的死亡事件发生。
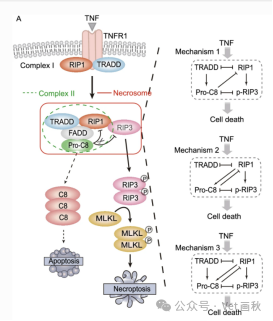
坏死小体关键蛋白 RIP1 和 RIP3 在坏死性凋亡中的作用机制
在坏死小体中,存在两个至关重要的蛋白——RIP1 和 RIP3。它们的形成过程不仅涉及结构改变,更重要的是发生磷酸化修饰,这是因为二者的活性结构域具有高度相似性,就如同两个功能相似的 “小机器零件”。这种磷酸化过程能够促使 TNFR 复合物 II 更加稳定,与此同时,激活 RIP3 下游的调控信号通路,就像启动了一条复杂的 “信号传导链”。RIP1 与 RIP3 相互作用并通过二聚化形成坏死小体,进而诱导坏死性凋亡的发生。在大鼠视网膜不存在缺血 - 再灌注损伤的情况下,细胞死亡则依旧按照常规的凋亡或坏死方式进行,如同细胞遵循着既定的 “死亡程序”。然而,当 RIP3 过度表达时,不仅能够检测到高浓度的活性氧产生,而且还会增强对 NF - κB 蛋白的调控作用,尽管这种调控能力相较于 RIP1 明显较弱。诸多疾病正是通过这种坏死性凋亡的方式对细胞死亡进行调控,这表明 RIP3 可能与疾病形成过程中其自身的活性存在密切关联,并且 RIP3 具备控制体内细胞向坏死性凋亡转化的能力,如同一个 “细胞命运的调控开关”。除了依赖 RIP1 和 RIP3 的活性外,坏死性凋亡(包括 RIP1 活性)还能够直接或间接参与 RIP3 在 Ser161 磷酸化位点的磷酸化过程。研究表明,在小鼠细胞中,病毒感染主要是由 RIP3 调控的坏死性凋亡所主导,而非 RIP1 调控。随着研究的不断深入,越来越多的证据显示,某些类型的坏死实际上是由严格控制的内在细胞程序所介导的,这种新型的坏死形式如今被定义为坏死性凋亡。
一氧化氮合酶(NOS)系统:
氧化氮合酶(NOS)的类型与基本功能
在生物体内,已明确识别的一氧化氮合酶(NOS)存在三种类型,即神经型一氧化氮合酶(nNOS)、内皮型一氧化氮合酶(eNOS)和诱导型一氧化氮合酶(iNOS)。这三种类型的 NOS 均具备将 L - 精氨酸转化为 L - 瓜氨酸的能力,在此过程中会产生一氧化氮(NO)。NO 作为一种重要的生物活性分子,凭借其抗氧化和抗炎特性发挥着对机体的保护作用,犹如机体内部的 “防御卫士”,在维持细胞正常生理功能和内环境稳定方面扮演着不可或缺的角色。
缺氧状态下NOS的变化及影响
然而,当机体处于缺氧状态时,NOS 会发生显著变化,转变为一种特殊状态 ——NOS 解偶联。在这种状态下,NOS 不再产生具有保护作用的 NO,反而开始产生一系列具有高度活性的分子 —— 活性氧(ROS),这些 ROS 如同 “捣乱分子”,会引发缺血 - 再灌注损伤。NOS 的结构较为复杂,其包含两个关键结构域:一个是加氧酶结构域,此结构域中含有血红素和四氢生物蝶呤(BH4),并且能够与精氨酸相结合;另一个是还原酶结构域,其中含有黄素(FAD 和 FMN),并具备与 NADPH 结合的能力。四氢生物蝶呤(BH4)作为一种重要的辅因子,在 NO 的产生过程中起着关键的调控作用。它是由鸟苷 5' - 三磷酸(GTP)经过一系列复杂的生化反应,包括 GTP 环化水解酶 I、醌式二氢生物蝶呤(BH2)和墨蝶呤等中间产物逐步合成而来。BH4 的浓度水平直接影响着 eNOS 的活性,进而对 NO 的产生量进行精细调控,如同一个 “生物活性调节器”,确保 NO 的产生维持在适宜的水平。
缺血-再灌注损伤中 NOS 相关的变化及机制
在缺血 - 再灌注损伤的情境下,氧化应激反应加剧,如同一场 “内部风暴”,会导致 BH4 被氧化,进而使组织内 BH4 的水平显著降低。当 BH4 与 NOS 的比值降低时,会引发 NOS 与超氧阴离子发生解偶联现象,这一过程如同 “链条断裂”,破坏了正常的生理功能关联,最终导致细胞面临死亡风险。缺血 - 再灌注损伤过程中 ROS 的产生机制是一个多因素且错综复杂的过程。在缺血阶段,多个酶系统如黄嘌呤氧化酶系统、NADPH 氧化酶系统、线粒体电子传递链以及解偶联的 NOS 系统等均会产生 ROS。这些 ROS 如同 “有害物质的堆积”,会在细胞内不断积累,从而削弱抗氧化剂的防御效能,使细胞处于氧化应激的 “高压环境” 之下。当缺血组织的血液供应恢复后,如果氧化应激的程度达到一定严重程度,细胞将会遭受损伤,甚至发生死亡,这一系列过程构成了缺血 - 再灌注损伤诱导细胞死亡的复杂机制。
线粒体损伤(线粒体功能障碍)
线粒体的结构与功能及其相关代谢机制
缺血对电子传递链复合物的影响
短时间缺血:要是缺血的时间比较短呀,就好像是给电子传递链复合物来了个 “小刺激”,会让它们的电负性增加,还会出现电子泄漏的情况。不过,好在这个时候线粒体自身的抗氧化能力还没怎么受影响,就像它自带的 “防御盾牌” 还能正常发挥作用一样,所以即便之后再灌注的时候产生了那些活性氧(ROS,就像是一群 “捣蛋分子”),也还是能被线粒体的抗氧化系统给 “消灭” 掉。
长时间缺血:但要是缺血时间一长呀,那电子传递链复合物可就 “遭了殃” 了。经过 60 分钟的热缺血之后呢,所有这些复合物的活性都会下降,它们的亚基就像建筑的 “零部件” 一样,会出现结构上的损伤。而且呀,这里面复合物 I 和复合物 III 对缺血带来的这种损伤好像更加敏感,就像它们俩是比较 “脆弱” 的环节。另外呢,腺嘌呤核苷酸转位酶(ANT)这个负责转运工作的 “小助手”,它的转运能力也会因为酰基辅酶 A 酯越堆越多而变弱了。更麻烦的是,在大鼠肝脏遭遇冷缺血损伤之后呀,复合物 V(ATP 合酶)这个 “关键设备” 在分子层面上都出现损伤了。这些受了伤的电子传递链复合物呢就更容易出现电子泄漏的问题了,而且这种情况在再灌注结束之后,还可能会持续挺长一段时间。
三磷酸腺苷(ATP)耗竭相关情况
在缺血的时候,ATP 会发生水解,这一水解可不得了,就像引发了一连串的 “连锁反应”,会让游离的无机磷酸盐变多了,而这一变多,又会让细胞膜的通透性增大。同时,ATP 水解再加上无氧糖酵解这两个过程一起 “发力”,会让细胞里面的 pH 值降低,不过这个 pH 值降低倒也不完全是坏事,它在缺血期间还能像一个 “保护罩” 一样,起到保护细胞、让细胞不至于那么容易死亡的作用。要是热缺血的时间拖得很长,那就又有新问题了,会让和电子传递链里复合物 I(也就是 NADH 脱氢酶)或者复合物 II(琥珀酸脱氢酶)相关联的铁硫蛋白慢慢变少,就好像这些蛋白在慢慢 “消失” 一样,然后亚铁离子(Fe²⁺)就被释放出来了。这个亚铁离子在再灌注的时候可就成了个 “关键角色” 了,它能把过氧化氢(H₂O₂)还原,然后产生那些活性氧(ROS),对整个过程产生很重要的影响。
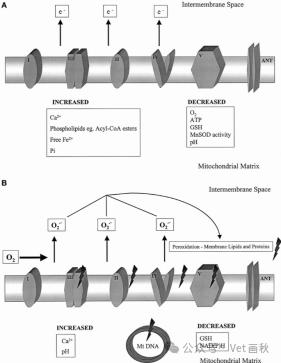
抗氧化水平变化情况
缺血这件事,还会对线粒体的抗氧化防御系统造成破坏,就好像把线粒体的 “防御城墙” 给弄出了漏洞一样,这样一来,细胞就变得更容易受到氧化应激的 “攻击” 了。线粒体里有一些谷胱甘肽酶,像谷胱甘肽过氧化物酶(GSHpx),它的活性在缺血的时候好像还相对比较稳定,还能勉强维持着。但是线粒体里的谷胱甘肽(GSH)水平就没那么幸运了,不管是在热缺血的时候,还是大鼠肝脏被冷藏的过程中,它的含量都有可能下降。而且线粒体锰超氧化物歧化酶(MnSOD)好像比细胞质里的铜锌超氧化物歧化酶(Cu, ZnSOD)更经不起缺血的 “折腾”,在全脑缺血 60 分钟之后,锰超氧化物歧化酶(MnSOD)的活性就明显变低了。不过这里面有个挺有意思的事儿,要是把锰超氧化物歧化酶的基因导入进去,还能起到保护大鼠肝脏的作用,让肝脏免受氧化应激带来的损伤。
缺血期间离子变化及其他影响
在缺血的时候,细胞里面的情况也变得挺复杂的。因为细胞质里钠离子(Na⁺)的含量升高了,就导致钠钾泵(Na⁺/K⁺泵)这个维持细胞内外离子平衡的 “小机器” 没办法正常工作了,就像它被 “卡住” 了一样。这一 “卡住” 可就引发了后续一系列变化,细胞质里的钙离子(Ca²⁺)跟着就变多了,然后线粒体里的钙离子(Ca²⁺)也跟着增加,这么一来呀,线粒体内膜的通透性就变大了。还有个新发现哦,最近有研究表明,线粒体在调节缺氧诱导因子 1 的表达方面还起着作用呢。这个缺氧诱导因子1就像是一个 “指挥官”,它是一种转录因子,能够控制好多个对缺氧有反应的基因的激活情况。
再灌注相关情况及影响
线粒体在再灌注氧化损伤中的作用体现
在研究缺血再灌注损伤的那些实验里,已经发现线粒体在氧化损伤这件事儿上起着不小的作用呢。比如说在肝脏缺血再灌注损伤的模型中,通过使用专门针对电子传递链的抑制剂做实验,就确定了线粒体是产生过氧化氢的一个重要 “源头”。把分离出来的肝细胞放在缺氧然后又复氧的环境里时,这些肝细胞会产生超氧阴离子自由基(O₂・⁻),还会释放乳酸脱氢酶呢。有意思的是,要是用别嘌醇(它可是一种能抑制黄嘌呤氧化酶的东西)去处理这些细胞,发现对这个产生超氧阴离子自由基(O₂・⁻)的情况没啥影响,这就说明线粒体才是产生超氧阴离子自由基(O₂・⁻)的主要 “源头” 。
还有,要是把线粒体单独分离出来,然后让它们也经历缺氧再复氧的过程,这时候线粒体的呼吸作用就会变弱了,就好像它们变得有点 “没力气” 了一样,而且这还和线粒体里面蛋白质被氧化损伤以及能检测到脂质过氧化的标志物丙二醛这些情况是相关联的。不过,要是在缺氧/复氧之后,用那些可溶性的抗氧化剂来 “照顾” 一下线粒体,就像给它们 “补充能量” 一样,还是有助于让线粒体继续保持活跃的呼吸作用的。
长时间缺血后再灌注面临的问题及影响
要是缺血的时间拖得很长,那身体里原本用来对抗 ROS 的抗氧化系统可就 “吃不消” 了,它本身已经受到了损伤,就没办法很好地应对这时候过量产生的活性氧(ROS)了。就好比一个 “防御部队”,已经自身难保了,自然没法有效地抵挡 “敌人” 了。这时候,因为锰超氧化物歧化酶(MnSOD)的活性不足,大量产生的超氧阴离子自由基(O₂・⁻)就会发生反应,生成像过氧亚硝酸盐这样很强的氧化剂,这个过氧亚硝酸盐呀,就是超氧阴离子自由基和线粒体一氧化氮相互反应后产生的。
另外,要是谷胱甘肽(GSH)的量不够了,那过氧化氢(H₂O₂)就会越积越多,这个过氧化氢虽然本身不是自由基那种 “活泼” 的物质,但它可以通过一种叫芬顿反应的过程(在这个反应里铁就像是一个 “催化剂”,能起到推动作用),形成那些毒性更强的物质,比如说羟基自由基。有研究还发现呢,在心脏再灌注之后没多久,线粒体里 90% 的氧气居然是以羟基自由基的形式存在的,这可不得了。
而且,像肿瘤坏死因子(TNF)这样的促炎细胞因子,在缺血 / 再灌注之后,它的量可能会增多,就像被 “激活” 了一样,然后还会刺激线粒体去产生活性氧(ROS)。经过研究发现,这个 TNF 产生活性氧(ROS)的过程,是由它在信号通路里产生的神经酰胺来介导的,就好像是通过一条特定的 “传导链条” 来实现的。
再灌注对线粒体及细胞各方面的影响
急性的氧化应激一来,就会对线粒体的呼吸作用起到抑制的效果,那些活性氧(尤其是羟基自由基和过氧亚硝酸盐)就像一群 “破坏分子” 一样,会去和线粒体以及线粒体外的各种结构(像蛋白质、脂质还有DNA 这些)发生反应,然后对它们造成损伤。电子传递链复合物里的蛋白质一旦受损了,就会进一步影响到三磷酸腺苷(ATP)的产生,就好像一条 “生产线” 上的关键 “零件” 坏了,整个生产流程都会受影响一样,而且还会形成蛋白质和蛋白质之间相互连接在一起的情况,这种情况又会让电子更容易泄漏出去,就像 “漏洞” 变得更大了一样。
活性氧(ROS)还会和磷脂发生反应,它们反应之后产生的一些东西,比如说 4 - 羟基壬烯醛,这个物质,会进一步去损害细胞膜,还会影响到线粒体内膜的通透性,就好像是把内膜原本的 “防护屏障” 给破坏了一样。这么一来,电子传递就会变得更不顺畅了,过氧化氢(H₂O₂)的产生也会跟着变多了。除此之外,吡啶核苷酸要是被氧化了,也会改变细胞膜的通透性,还会让氧化磷酸化这个过程变得 “脱节”,导致三磷酸腺苷(ATP)没办法正常产生了。
再灌注之后呀,钙离子(Ca²⁺)可能会大量地进入线粒体里面,这就好比一下子涌进了很多不该进来的 “访客” 一样,这么多钙离子进来,会抑制氧化磷酸化这个过程,还会让线粒体内膜的通透性变大。细胞器里面的 pH 值在缺血的时候是下降的,等到再灌注之后呀,它又会恢复到正常的水平,可别小看这个变化,它可能会通过影响线粒体膜的通透性,来让细胞死亡的速度加快。对冷藏的大鼠肝脏进行再灌注之后,会发现谷胱甘肽(GSH)的水平降低了,同时线粒体羟基过氧化物酶的量在大鼠肝脏移植 30 分钟后还增加了。而谷胱甘肽(GSH)要是不足了,那对三磷酸腺苷(ATP)的产生、内膜的通透性以及细胞的活力都会产生影响的。
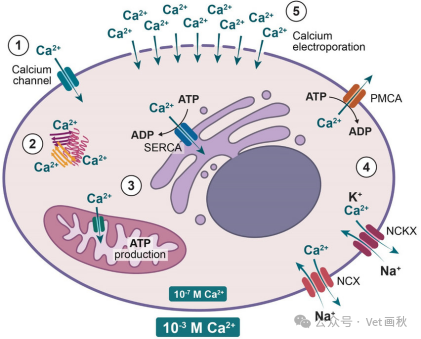
钙超载及其引发的系列病理生理变化
在缺血 - 再灌注阶段,细胞内常出现钙超载现象,且多在再灌注期发生,其主要原因是钙内流增多,致使细胞内钙离子数量超出正常水平,而钠钙交换异常、反向转运增强是其中关键情况。
钙超载的激活机制
直接激活机制
在缺血时,细胞内三磷酸腺苷(ATP)的生成量明显减少,ATP 对细胞而言如同为众多 “小机器” 提供动力的 “燃料”,其减少会使维持细胞内外离子平衡的钠钾泵运转受阻,进而导致细胞内钠离子(Na⁺)含量显著升高。到再灌注时,胞内高浓度的 Na⁺除激活钠泵外,还会激活钠钙交换蛋白的反向转运,使得更多的钙离子(Ca²⁺)进入细胞内,最终引发钙超载。
间接激活机制
缺血状态下,因细胞无法正常进行有氧呼吸,无氧酵解增强,产生的氢离子(H⁺)增多,致使组织间液及细胞内出现酸中毒。再灌注时,细胞外 H⁺浓度迅速下降,形成明显的 pH 梯度差,这一梯度差会激活细胞膜上的 H⁺ - Na⁺交换蛋白,使细胞内 Na⁺浓度再次增加,继发性激活钠钙交换蛋白反向转运,促使更多 Ca²⁺进入细胞。此外,蛋白激酶 C(PKC)活化后,可通过激活 H⁺ - Na⁺交换蛋白间接激活钠钙交换蛋白的反向转运,进一步加重钙超载情况。
儿茶酚胺增加的影响
儿茶酚胺在这一过程中也起着重要作用,它一方面作用于 α 受体,促进肌质网钙释放通道开放以及 H⁺ - Na⁺交换;另一方面作用于 β 受体,增加 L 型钙通道的开放程度,相当于为钙离子进入细胞开辟了多条通道,推动钙超载情况加剧。
生物膜损伤相关影响
细胞膜损伤方面:生物膜损伤是导致钙超载的重要因素之一。在该过程中,细胞膜对 Ca²⁺的通透性增加,再灌注时,细胞外 Ca²⁺顺着浓度差大量涌入细胞内。并且,细胞内 Ca²⁺增多会激活磷脂酶,促使膜磷脂降解,进一步增大细胞膜对 Ca²⁺的通透性,形成恶性循环。同时,再灌注时生成的大量自由基会攻击细胞膜,加重细胞膜损伤,也使得钙超载愈发严重。
肌浆网膜与线粒体膜损伤方面:若钙泵出现功能障碍,肌浆网对 Ca²⁺的摄取量会减少,导致细胞内 Ca²⁺浓度升高。大量钙盐沉积于线粒体,会造成线粒体氧化磷酸化障碍,使 ATP 生成减少,细胞膜和钙泵因能量供应不足,无法有效控制细胞内钙离子量,进而加重钙超载。而且,自由基损伤及膜磷脂降解也会伤害线粒体膜,使其产生 ATP 的能力进一步下降,持续加重钙超载情况。
钙超载对自由基产生的影响
Ca²⁺促进 XD - XO 转化导致自由基增加
XD - XO 转化机制基础:黄嘌呤脱氢酶(XD)和黄嘌呤氧化酶(XO)参与细胞内嘌呤代谢过程。正常时,细胞内主要是 XD 在发挥作用,它在代谢次黄嘌呤等底物时,优先以 NAD⁺作为电子受体,过程平稳,不会大量产生活性氧(ROS)自由基。但在缺血 - 再灌注损伤等特殊病理生理情况下,XD 会受多种因素影响转化为 XO,细胞内钙超载就是重要触发因素。
Ca²⁺对 XD - XO 转化的具体作用:当细胞内出现钙超载,过多的 Ca²⁺会结合到 XD 上特定的钙结合位点(由酶蛋白特定氨基酸残基构成),引发 XD 发生如有限蛋白水解、关键活性调节区域空间结构改变等一系列变化,使其转变为氧化活性更强的 XO。同时,细胞内高浓度 Ca²⁺还会激活某些蛋白激酶,对 XD 进行修饰(如磷酸化),改变其稳定性和活性,促使其向 XO 转化。
XO 产生自由基的过程及影响:XD 转化为 XO 后,XO 在处理次黄嘌呤和黄嘌呤时,电子受体变为氧气(O₂)。在此过程中,XO 将电子传递给 O₂,使 O₂ 通过单电子还原生成超氧阴离子自由基(O₂・⁻),该自由基还可经其他反应转化为过氧化氢(H₂O₂)等活性氧自由基。这些自由基会攻击细胞膜不饱和脂肪酸引发脂质过氧化反应,破坏细胞膜完整性和流动性;攻击细胞内蛋白质使其失活、变性,扰乱酶促反应和信号传递过程;对核酸(DNA 和 RNA)造成氧化损伤,导致基因突变、影响基因转录和翻译等,严重干扰细胞正常生理功能,促使细胞走向损伤甚至凋亡。
Ca²⁺导致线粒体功能受阻进而使自由基增加
线粒体正常生理功能与自由基产生的关联:线粒体作为细胞的 “能量工厂”,通过氧化磷酸化过程生产 ATP。在此过程中,电子传递链(ETC)起着关键作用,电子在 ETC 各复合物间依次传递,最终与氧气结合生成水,同时释放能量建立电化学梯度,驱动 ATP 合酶合成 ATP。正常情况下虽有少量电子泄漏生成少量活性氧自由基(如超氧阴离子自由基),但细胞内自身的抗氧化防御系统(如超氧化物歧化酶、谷胱甘肽过氧化物酶等)可及时清除,维持氧化还原平衡。
Ca²⁺对线粒体功能的影响及自由基生成增加机制
钙超载影响线粒体钙稳态及膜电位:细胞内钙超载时,过量 Ca²⁺会大量涌入线粒体。线粒体虽有摄取 Ca²⁺的能力,但其内膜转运蛋白摄取过多 Ca²⁺后,会破坏线粒体精密的钙稳态调节机制,改变线粒体膜电位使其去极化,进而干扰电子传递链复合物正常运转,破坏质子梯度维持,导致氧化磷酸化过程受干扰,ATP 生成减少。
对电子传递链的影响:大量 Ca²⁺在线粒体积累后,会与电子传递链复合物中参与电子传递的金属离子(如铁、铜等)结合位点相互作用,改变复合物构象和活性,使电子传递过程紊乱,电子泄漏加剧,更多电子提前与氧气反应生成超氧阴离子自由基等活性氧自由基。同时,钙超载引起的膜电位变化也会改变电子传递动力和效率,进一步促进电子泄漏和自由基产生。
对线粒体抗氧化防御系统的削弱:线粒体功能受干扰后,其内部负责维持谷胱甘肽(GSH)还原态等抗氧化防御相关酶因 ATP 减少、内部环境紊乱而无法正常工作,不能及时有效清除不断增加的自由基,致使自由基在细胞内积累,氧化应激损伤加重。
线粒体通透性转换孔(mPTP)开放相关影响:钙超载还是线粒体通透性转换孔(mPTP)开放的重要诱导因素,mPTP 开放使线粒体膜通透性急剧增大,一方面导致线粒体基质内细胞色素 c 等物质释放到胞质,影响细胞凋亡调控机制;另一方面使细胞外物质更易进入线粒体,扰乱其内部代谢和离子平衡,加剧自由基产生,同时降低其自身清除自由基能力,最终导致细胞内氧化还原状态严重失衡,对细胞造成极大伤害。
总之,细胞内钙超载通过促进 XD - XO 转化以及干扰线粒体功能这两个主要途径,使细胞内自由基大量增加,打破氧化还原平衡,引发一系列病理生理变化,是缺血 - 再灌注损伤中导致组织损伤的重要环节。
中性粒细胞在缺血/再灌注损伤中的核心作用
在缺血/再灌注(IR)损伤过程中,中性粒细胞起着极为重要的作用,是引发一系列连锁反应的关键因素。
黄嘌呤氧化酶(XO)、血小板活化因子(PAF)和活性氧(ROS)能引发趋化作用,吸引中性粒细胞聚集,进而出现中性粒细胞浸润情况。白细胞浸润,尤其是中性粒细胞浸润,是缺血 / 再灌注级联反应的关键环节。
中性粒细胞表面的细胞黏附分子与内皮细胞上的配体相结合,会引发一系列事件,最终使白细胞从微血管渗出,导致组织损伤。再灌注时,大量常驻或浸润的中性粒细胞产生氧化剂并释放蛋白水解酶,是造成大部分组织损伤的原因。活化的中性粒细胞通过呼吸爆发期间合成活性氧、释放内源性蛋白水解酶以及对毛细血管造成物理性阻塞等方式损伤组织。
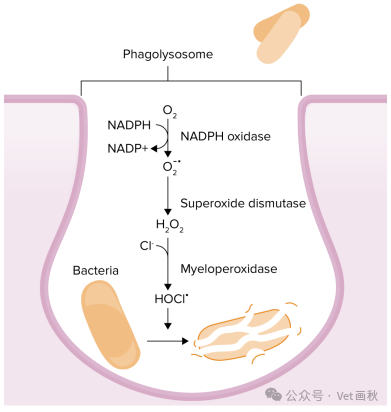
在猫肠道黏膜再灌注后,预先使用别嘌醇(一种黄嘌呤氧化酶抑制剂)、超氧化物歧化酶、过氧化氢酶和去铁胺可阻止中性粒细胞浸润,有力证明了活性氧、中性粒细胞浸润与后续组织损伤之间存在紧密关联。此外,采用黄嘌呤氧化酶抑制剂、减少中性粒细胞数量、应用二甲基亚砜(羟基自由基清除剂)或针对 CD18 中性粒细胞黏附分子的单克隆抗体进行干预后,缺血 / 再灌注所致的微血管通透性增加情况会有所减轻。多项研究表明,中性粒细胞介导了缺血 / 再灌注后大部分黏膜和微血管损伤。
中性粒细胞与内皮细胞黏附及渗出的多步骤过程
微血管对缺血 / 再灌注相当敏感,中性粒细胞与内皮细胞黏附性增加会导致缺血 / 再灌注时出现微血管损伤。
中性粒细胞从微血管渗出涉及一系列复杂的多步骤事件,包括栓系 / 滚动、牢固黏附、跨膜迁移以及趋化作用,其中选择素和整合素参与其中。
在血流状态下,中性粒细胞通过活化内皮细胞上的 E - 选择素和 P - 选择素与白细胞 L - 选择素同各种膜糖蛋白相互作用而发生栓系和滚动。滚动主要由内皮细胞的活化引发,早期由组成性表达的内皮细胞 P - 选择素和中性粒细胞 L - 选择素介导,E - 选择素较晚参与,因其需从头合成。
一旦中性粒细胞通过选择素相互作用与内皮细胞栓系在一起,中性粒细胞整合素受体内皮细胞表达的血小板活化因子、趋化因子或局部分泌的趋化物质激活,激活后其对内皮细胞配体 —— 细胞间黏附分子 - 1(ICAM - 1)和细胞间黏附分子 - 2(ICAM - 2)的亲和力增加。
中性粒细胞黏附到内皮细胞上后,会在内皮细胞表面移动,然后穿过内皮细胞间连接渗出,并迁移至炎症部位。弹性蛋白酶或其他介质破坏细胞间连接的完整性会使中性粒细胞迁移更容易。中性粒细胞的募集可通过内吞作用去除内皮细胞上的 E - 选择素和 P - 选择素,以及通过一种膜蛋白酶从中性粒细胞上裂解 L - 选择素等机制终止。
中性粒细胞黏附的不同阶段及相关机制
研究表明,在缺氧及复氧后,中性粒细胞与内皮细胞的黏附存在早期(第 1 阶段)和后期(第 2 阶段)两个阶段。
早期阶段由血小板活化因子和黄嘌呤氧化酶衍生的过氧化氢介导。第 1 阶段中性粒细胞黏附模式是因组成性表达的细胞间黏附分子 - 1、可快速动员的预先形成的 P - 选择素库以及中性粒细胞表面 β2 - 整合素表达上调所致。
核转录因子(NFκB)在第 2 阶段中性粒细胞黏附时内皮细胞黏附分子的上调过程中起重要作用。NFκB 通常存在于细胞质中,需转移到细胞核才能引发相应反应。第 2 阶段依赖 E - 选择素的中性粒细胞黏附模式与由 NFκB 介导的黏附蛋白转录依赖性上调相符。抑制 NFκB 激活可阻止 E - 选择素表面表达增加,这与中性粒细胞对内皮细胞单层黏附的减少直接相关。
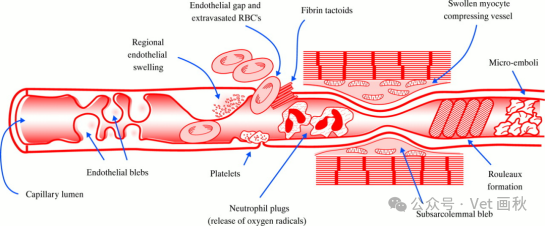
再灌注期间的恶性循环及相关炎症介质
再灌注期间会出现恶性循环,持续的中性粒细胞趋化和活化会导致更多活性氧形成、内皮损伤以及毛细血管堵塞。
通过活体显微镜观察发现,猫肠系膜再灌注 10 分钟后,中性粒细胞对静脉内皮出现最大黏附现象。在其他物种和器官中也有类似时间变化情况,如马的大肠在低流量缺血 3 小时后恢复血流 10 分钟时也会出现此类情况。
多项研究中,缺血 / 再灌注损伤部位的髓过氧化物酶(MPO)活性增加,证明了中性粒细胞聚集现象的发生及其对损伤的促成作用。髓过氧化物酶活性与丙二醛浓度(即脂质过氧化的标志物)的相关性表明,脂质过氧化损伤发生在中性粒细胞衍生的活性氧之后。也有可能髓过氧化物酶活性增加发生在细胞膜磷脂脂质过氧化过程中释放的磷脂衍生介质(如血小板活化因子)增加之后。
缺血/再灌注期间产生的炎症介质包括肿瘤坏死因子 α(TNFα)、白细胞介素 - 1β(IL - 1β)、血小板活化因子、补体以及趋化因子,它们都是强效的趋化物质,会促使中性粒细胞扣押增加。虽然活化的中性粒细胞自身能释放肿瘤坏死因子 α 和白细胞介素 - 1β,但这些细胞因子主要由常驻巨噬细胞和肥大细胞释放。肿瘤坏死因子 α 可诱导中性粒细胞黏附和脱颗粒,刺激 NADPH 氧化酶(导致活性氧合成),并增强白细胞介素 - 2 受体以及内皮细胞上细胞间黏附分子 - 1 的表达。
中性粒细胞脱颗粒会增加氧化应激,增加组织中弹性蛋白酶和胶原酶的活性,并促进类花生酸合成。弹性蛋白酶是中性粒细胞释放的导致组织损伤的主要蛋白水解酶。黄嘌呤氧化酶衍生的活性氧启动中性粒细胞的募集,随后中性粒细胞被激活并释放活性氧,导致器官损伤加剧。循环白细胞会导致微血管通透性增加,常驻的间质粒细胞则会导致与缺血 / 再灌注相关的胃肠道黏膜通透性增加。
无复流现象及中性粒细胞在其中的作用
“无复流现象” 用于描述在解除血管阻塞后,某一组织区域血流量减少或缺失的情况。该概念最初由Majno等人提出,他们对兔子进行不同时长的脑缺血实验,发现短暂(2.5 分钟)缺血后血流能够恢复,但缺血时间较长时血流则无法恢复。该模型在其他缺血动物模型以及皮肤、骨骼肌和肾脏等其他器官中也进行了重复实验。
Kloner等人在犬近端冠状动脉阻塞模型中研究心脏血管的电子显微镜样本,观察到内皮细胞肿胀、内皮突出以及血小板和纤维蛋白血栓,认为这些是导致动物出现无复流现象的原因。实验研究表明,缺血时间越长越易出现无复流现象。
中性粒细胞在无复流现象中可能起着关键作用。黏附在内皮细胞上的中性粒细胞造成损伤,导致内皮肿胀,进而吸引更多中性粒细胞聚集,形成恶性循环,使更多中性粒细胞被吸引到该区域。多项研究显示,通过减少中性粒细胞或血小板的数量,无复流现象会有所减轻。
血管损伤导致内皮剥脱或回缩后,血小板会迅速黏附到暴露的内皮下层,随后通过启动依赖选择素和整合素的白细胞与表面结合血小板的黏附作用,招募更多的中性粒细胞。黏附在血小板上的聚集的中性粒细胞可能会促进纤维蛋白沉积以及随后的血栓形成。由于在阻塞消除后往往先有一段正常血流期,随后血流量又会减少,因此提出了 “复流减少” 这一术语。

